INTRODUCTION
SUMMARY
The heavy-chain diseases (HCDs) are B-cell lymphoplasmacytic proliferative disorders in which neoplastic cells produce monoclonal immunoglobulins (Ig) consisting of truncated heavy chains (HCs) without attached light chains. The complex abnormalities of HCD proteins and the usual lack of normal light chains are a result of several distinct gene alterations, including somatic mutations, deletions, and insertions. HCDs involving the three main immunoglobulin classes have been described: α-HCD is the most common and has the most uniform presentation; γ– and μ-HCDs have variable clinical presentations and histopathologic features. The diagnosis is established from immunofixation of serum, urine, or secretory fluids in the case of α-HCD or by immunohistologic analysis of the proliferating lymphoplasmacytic cells in nonsecretory disease. Treatment of α-HCD consists of antibiotics. If there is no response to antibiotics or if aggressive lymphoma is diagnosed, chemotherapy is indicated. Treatment of γ– and μ-HCDs depends on the underlying clinicopathologic features rather than on the presence of the abnormal protein. Table 110–1 summarizes the features of the HCDs.
Acronyms and Abbreviations
CH1 (2, 3, 4), constant region 1 (2, 3, 4); D, diversity; HC, heavy chain; HCD, heavy-chain disease; Ig, immunoglobulin; IPSID, immunoproliferative small intestinal disease; J, joining; V, variable.
| Types of Heavy-Chain Diseases | |||
|---|---|---|---|
| Features | α | γ | μ |
| Year described | 1968 | 1964 | 1969 |
| Incidence | Rare | Very rare | Very rare |
| Age at diagnosis | Young adult (<30 years) | Older adult (60–70 years) | Older adult (50–60 years) |
| Demographics | Mediterranean region | Worldwide | Worldwide |
| Structurally abnormal monoclonal protein | IgA | IgG | IgM |
| MGUS phase | No | Rarely | Rarely |
| Urine monoclonal light chain | No | No | Yes |
| Urine abnormal heavy chain | Small amounts | Often present | Infrequent |
| Sites involved | Small intestine, mesenteric lymph nodes | Lymph nodes, marrow, spleen | Lymph nodes, marrow, liver, spleen |
| Pathology | Extranodal marginal zone lymphoma (MALT or IPSID) | Lymphoplasmacytoid lymphoma | Small lymphocytic lymphoma, CLL |
| Associated diseases | Infection, malabsorption | Autoimmune diseases | None |
| Therapy | Antibiotics, chemotherapy | Chemotherapy | Chemotherapy |
γ-HEAVY-CHAIN DISEASE DEFINITION AND HISTORY
γ-Heavy-chain disease (HCD) is not a specific cytopathologic process; rather, it is a biochemical expression of a mutant B-cell clone. The disease should be considered a serologically determined entity with a variety of clinical and histopathologic features. It is defined by the recognition of monoclonal deleted gamma (γ) chains devoid of light chains.1
The first case of γ-HCD was described in 1964 by Franklin and colleagues,2 who observed a homogeneous band between γ– and β-globulin in an African American patient with generalized lymphadenopathy. Comparison of the proteins in the urine to those in the serum showed that they were the same, a suggestion of the presence of a low-molecular-weight serum γ-globulin, which then was shown to be a fragment of a γ-heavy chain (HC). Since this first description, more than 130 patients with γ-HCD have been described in the literature.3,4,5
EPIDEMIOLOGY
CLINICAL FEATURES
Originally, γ-HCD was considered to be a lymphomatous illness. However, γ-HCD has various clinical and pathologic features that can be divided into three broad categories, as described below.
Disseminated lymphoproliferative disease is present in most patients at the time of diagnosis, and various series have reported it in 57 percent to 66 percent of patients.4,5,6 In two different series,4,5 lymphadenopathy was present in 56 percent and 62 percent of patients, splenomegaly in 38 percent and 52 percent, and hepatomegaly in 8 percent and 37 percent at the time of diagnosis.
In approximately 25 percent of patients, the lymphoproliferative process is localized, which may be extramedullary or may involve only the marrow.4,5 Cutaneous involvement is the most frequently reported extramedullary presentation.4,5 Patients may present with extramedullary plasmacytoma of the thyroid or parotid gland or an oropharyngeal mass5 and hypertrophic spinal pachymeningitis.7
A proliferative lymphoplasmacytic disease is not apparent in 9 percent to 17 percent of patients. In most of these patients, an underlying autoimmune disorder has been reported. Autoimmune disorders with or without underlying lymphoid proliferation include rheumatoid arthritis, autoimmune cytopenias, systemic lupus erythematosus, Sjögren syndrome, myasthenia gravis, thyroiditis, and vasculitis.5
LABORATORY FEATURES
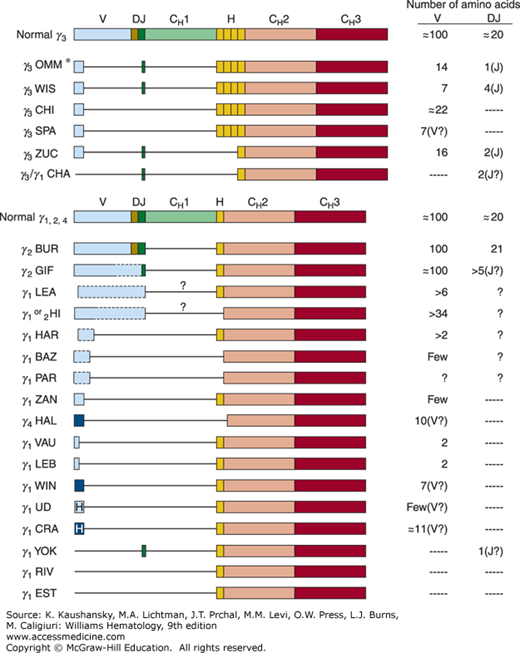
Most γ-HCD proteins are dimers of truncated HCs devoid of light chains. The molecular weight of the monomeric unit varies from 27,000 to 49,000. The length of the truncated γ chain varies, but usually it is one-half to three-fourths the length of the normal γ chain. Structural analysis of the defective monoclonal γ-HC of 23 patients with γ-HCD showed several characteristic features (Fig. 110–1). The proteins usually begin with a normal variable region. In most cases, this sequence is short and interrupted by a large deletion encompassing the remainder of the variable region, although four of the HCs shown in Fig. 110–1 appear to have retained most or all of their variable (V), diversity (D), and joining (J) sequences. In all γ-HCD proteins, the entire constant region 1 (CH1) domain is also deleted, with normal sequence beginning at the hinge or occasionally at the CH2 domain. CH1 is responsible for light-chain binding. In the absence of an associated light chain, the CH1 domain binds to heat shock protein 78 (HC binding protein), and the HC undergoes proteasomal degradation rather than secretion.
Figure 110–1.
Structure of various deleted γ-heavy-chain disease (HCD) proteins compared with that of normal chains.* Structure shown is a primary synthetic product synthesized by the HCD cells. Serum protein was modified after synthesis and did not contain any amino acids before the hinge.  , indicates heterogeneous amino acid sequences;
, indicates heterogeneous amino acid sequences;  , unusual and heterogeneous amino acid sequences;
, unusual and heterogeneous amino acid sequences;  , unusual amino acid sequences; boxes, coding regions; lines, deletions; dashed lines, likely structures for which sequence data are missing; ?, probable missing domain based on molecular weight and partial protein structure analysis; V, variable region; D, diversity segment; J, joining region; H, hinge region; CH1, CH2, CH3, constant regions of heavy chains. OMM,66 WIS,67 CHI,68 SPA,69 ZUC,70 CHA,71 BUR,72 GIF,73 LEA,74 HI,75 HAR,74 BAZ,76 PAR,77 ZAN,78 HAL,79 VAU,80 LEB,80 WIN,81 UD,82 CRA,83 YOK,84 RIV,85 EST.86
, unusual amino acid sequences; boxes, coding regions; lines, deletions; dashed lines, likely structures for which sequence data are missing; ?, probable missing domain based on molecular weight and partial protein structure analysis; V, variable region; D, diversity segment; J, joining region; H, hinge region; CH1, CH2, CH3, constant regions of heavy chains. OMM,66 WIS,67 CHI,68 SPA,69 ZUC,70 CHA,71 BUR,72 GIF,73 LEA,74 HI,75 HAR,74 BAZ,76 PAR,77 ZAN,78 HAL,79 VAU,80 LEB,80 WIN,81 UD,82 CRA,83 YOK,84 RIV,85 EST.86
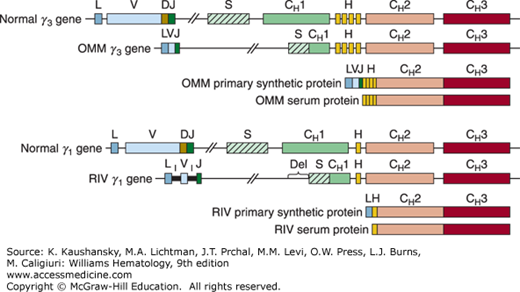
In two cases of γ-HCD (OMM and RIV), genomic sequence data are available (Fig. 110–2). The presence of large deletions in the switch/CH1 regions of these two γ-HCD genes explains why the corresponding HCD proteins lack CH1. Because the normal CH1 acceptor splice site is deleted, the donor splice of the leader, or the J region, is spliced directly to the next available functional acceptor splice site at the beginning of the hinge or CH2 domain.
Figure 110–2.
Structure of two genes coding for γ-heavy-chain-disease proteins compared with that of normal γ3 and γ1 genes. Boxes indicate coding regions;  , switch region;
, switch region;  , inserted noncoding sequence; lines, intervening (noncoding) sequences; L, leader region; V, variable region; D, diversity segment; J, joining region; S, switch region; H, hinge region; CH1, CH2, CH3, constant regions of heavy chains; I, inserted sequence; Del, deleted sequence. OMM,66 RIV.85
, inserted noncoding sequence; lines, intervening (noncoding) sequences; L, leader region; V, variable region; D, diversity segment; J, joining region; S, switch region; H, hinge region; CH1, CH2, CH3, constant regions of heavy chains; I, inserted sequence; Del, deleted sequence. OMM,66 RIV.85
The serum protein electrophoretic pattern is extremely variable. A monoclonal peak is detected in 60 percent to 86 percent of patients.4,5 When present, it is most commonly in the β1 or β2 region. The median value of the monoclonal spike at diagnosis in 19 patients was 1.59 g/dL (range: 0.40 to 3.91 g/dL).5 The diagnosis is established by immunofixation of the serum and a concentrated urine specimen. A modified immunoselection technique for the diagnosis of HCD has been described.8 In one case of γ-HCD, low concentrations of free HCs in serum were detected by capillary zone electrophoresis coupled with immunosubtraction.9 A heavy/light chain assay to identify truncated immunoglobulin (Ig) HCs was used on serum samples from 15 patients with known γ-HCD. By this method, 20 percent of these patients were shown to also have small amounts of monoclonal free light chains.10 The amount of HCD protein in the urine usually is small (<1 g/24 h) but may reach 20 g/24 h. An occasional patient with Bence Jones proteinuria has been described.5
No standard has been established to identify the subclass of the HC fragment. In reported cases in which the HC fragment subclass has been studied, different methods have been used, ranging from Ouchterlony in the earlier cases to indirect immunofluorescence staining, immunoblotting, amino acid sequence, immunoselection, and enzyme-linked immunosorbent assay.11 IgG subclass distribution shows a lower-than-expected incidence of IgG2. The most common subclass is IgG1, which occurs in 65 percent of cases. IgG3 has been identified in 27 percent of patients, IgG4 in 5 percent, and IgG2 in 3 percent.4 Although biclonal gammopathy has been reported in 1 percent to 8 percent of all patients with serum monoclonal components, the association between γ-HCD and another monoclonal protein is much higher. In a series of 23 patients with γ-HCD, 7 percent had an IgM-λ intact monoclonal Ig.5 No association between γ-HCD and monoclonal IgA has been described, although the IgG-IgA association was the most frequent in several series of biclonal gammopathies. One patient described in the literature was unique in that the serum contained two deleted γ chains of different subclasses (IgG1 and IgG2).12
Anemia is frequent. It is usually normochromic, normocytic, and moderate. Coombs-positive autoimmune hemolytic anemia has been reported in several cases and may be associated with thrombocytopenia (Evans syndrome). The total and differential leukocyte counts are usually normal. Lymphocytosis may occur, and an occasional patient presents with chronic lymphocytic leukemia. In some cases, rare plasmacytoid lymphocytes or plasma cells have been noted in the blood. Plasma cell leukemia has been reported in two patients.13,14
Marrow aspirates and biopsy specimens often show an increase of plasma cells, lymphocytes, or plasmacytoid lymphocytes, similar to the marrow findings in Waldenström macroglobulinemia. The typical marrow features of myeloma or chronic lymphocytic leukemia are rare. An unusual concurrence of T-cell large granular lymphocytic leukemia with γ-HCD has been reported.15 Marrow changes consistent with a myeloproliferative neoplasm have been noted in a few patients.5
Bone lesions are rare in γ-HCD. Cytogenetic studies have seldom been reported. No unique abnormalities or characteristics of lymphoma have been found.
In contrast to α-HCD, γ-HCD has no consistent morphologic pattern and a variety of underlying lymphoproliferative disorders have been described.16 The most frequent histopathologic finding is a pleomorphic malignant lymphoplasmacytic proliferation in marrow and lymph nodes. These lymphocytoid plasma cells express pan–B-cell markers and cytoplasmic γ-HC without light chains and are negative for CD5 and CD10.17
Lymphoma without any consistent morphologic type was diagnosed in 18 (38 percent) of 47 patients in whom lymph nodes were examined. A lymphoplasmacytic proliferation was present in 36 percent and hyperplastic nodes and plasmacytoma in 11 percent each; there was one case of Hodgkin lymphoma and one of probable Hodgkin lymphoma.6 Plasmacytic infiltration may be found in the salivary glands or thyroid.4,5,18
DIFFERENTIAL DIAGNOSIS
THERAPY
Because γ-HCD is a heterogeneous condition, the choice of therapy depends on the clinical findings. In an asymptomatic patient with a monoclonal γ-HC, no therapy is indicated. Any associated autoimmune disease should be managed with standard therapy for that specific disease type. In symptomatic patients with a low-grade lymphoplasmacytic malignancy, a trial of chlorambucil may be beneficial. Melphalan and prednisone can be used, if the proliferation is predominantly plasmacytic. A trial of cyclophosphamide, vincristine, and prednisone with or without doxorubicin is reasonable for patients with evidence of a progressive lymphoplasmacytic proliferative process or high-grade lymphoma. One patient achieved a complete response after six courses of fludarabine.19 Successful treatment of γ-HCD with low-dose etoposide has been reported.20 CD20 expression has been analyzed in only seven cases and was detected in six of the seven, including one in which CD20 expression appeared transient.5,21,22,23 Rituximab monotherapy was given in two cases, resulting in clinical responses in both.5,21 In two other cases, a combination of rituximab with chemotherapy had an antitumor effect in lymphoplasmacytic-type γ-HCD.22,24 In localized extramedullary plasmacytomas treated with radiation4 or surgical removal (or both), complete clinical and serologic remission has been achieved.
COURSE AND PROGNOSIS
The clinical course of γ-HCD is extremely variable and ranges from an asymptomatic, benign, or transient process to a rapidly progressive neoplasm leading to death within a few weeks. Patients with the features of only a monoclonal gammopathy have remained clinically well for 2 to 7 years of followup.5,25 Spontaneous disappearance of the γ-HCD protein has been reported.4 The median duration of survival in a series of 23 patients was 7.4 years (range: 1 month to more than 21 years).5
The amount of serum γ-HCD protein usually parallels the severity of the associated malignant process. Disappearance of the monoclonal component from serum and urine associated with apparent complete response has been induced by chemotherapy,19 radiotherapy,4 or surgical removal of a localized process. In some instances, however, the γ-HCD protein does not vary in parallel with the associated process, and relapse can occur without the reappearance of the pathologic protein.4
α-HEAVY-CHAIN DISEASE DEFINITION AND HISTORY
α-HCD is a proliferative disorder of B-lymphoid cells involving the IgA secretory immune system, especially the gastrointestinal tract. It is defined by the recognition of internally deleted monoclonal α chains devoid of light chains.
In the first case description of α-HCD, in 1968 by Seligman and colleagues,26 an Arab woman had severe malabsorption resulting from a lymphoplasmacytic infiltrate in the small bowel. Since then, more than 400 cases have been reported.
EPIDEMIOLOGY
The majority of reported cases have been from northern Africa, Israel, and surrounding Middle Eastern countries. A common variable for patients with α-HCD is a low socioeconomic status. In a study of the distribution of monoclonal gammopathies in Tunisia published in 1990, 17 percent of 198 cases were attributed to α-HCD.27 In a later study of 270 cases observed between 1992 and 2000 at the university hospital of Sfax in Tunisia, only 2.2 percent were attributed to α-HCD,28 a finding that might be partially explained by improved socioeconomic conditions. Similarly, a persistent decrease in the incidence of immunoproliferative small intestinal disease (IPSID) since 1986 as a result of improving sanitation has been reported from Iran29 and Greece.30 α-HCD has a predilection for young adults. The prevalence of the disease is slightly higher in males than in females.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


