GROWTH FAILURE
Growth failure may be classified as follows: hypopituitarism; emotional deprivation; chromosomal defects; systemic illness (moderate to severe); metabolic diseases; and chondrodystrophies.
This classification is not all-inclusive but rather serves as a framework for discussing the approach to children with a failure to grow adequately. The major emphasis of this chapter is on hypopituitarism and emotional deprivation.
HYPOPITUITARISM
Hypothalamic and pituitary dysfunction (partial or complete) may be classified using a number of systems: congenital versus acquired; isolated, partial, or panhypopituitarism; transient versus permanent; idiopathic versus organic; and primary (pituitary) versus secondary (affecting releasing factors).
The outline shown in Table 18-2 is a convenient list with which to organize a differential diagnosis of hypopituitarism in infants and children. It is not all-inclusive but highlights major categories of pituitary insufficiency. The criteria for the diagnosis of the growth hormone (GH) deficiency of hypopituitarism are as follows: short stature; growth failure; no significant underlying illness; no evidence for emotional deprivation; delayed bone age; normal body proportions; low circulating levels of insulin-like growth factor-I (IGF-I); and subnormal responses to at least two stimuli for GH release.
 |
CONGENITAL HYPOPITUITARISM
Developmental Pituitary Disorders.
Developmental pituitary disorders may be so severe that they are incompatible with life. In some children, there is hypoplasia or absence of the pituitary gland, but in others, developmental anomalies of the hypothalamus appear to be responsible for the lack of pituitary development.3,4 Anencephaly and holoprosencephaly (including arrhinencephaly, holotelencephaly, cyclopia, and cebocephaly) usually are incompatible with life. This group of anomalies compose a spectrum of developmental anomalies associated with failure of complete midline cleavage of the embryonic fore-brain. The children either have no pituitary or a hypoplastic gland and have maldevelopment of the target organs. Atrophy of the adrenal glands always is present, and death usually occurs from adrenal insufficiency. A partial form of holoprosencephaly exists (Kallmann syndrome) in which patients have anosmia caused by the agenesis of the olfactory lobes, and hypogonadotropic hypogonadism secondary to failure of hypothalamic gonadotropin-releasing hormone secretion5 (see Chap. 16 and Chap. 115).
Absence or hypoplasia of the anterior pituitary is also an uncommon cause of hypopituitarism. The former usually is incompatible with life, but the latter has a spectrum ranging from severe to mild deficiency. The clinical presentation depends on the amount of functioning hypothalamic and pituitary tissue. The main features are severe hypoglycemia and shock because of adrenal failure.
The anterior pituitary lobe develops from an upward diverticulum of the primitive buccal cavity (Rathke pouch) in the nasopharynx and must migrate to its usual location within the sella turcica. There are a number of ectopic sites in which the gland may lodge, from the submucosa of the nasopharynx (pharyngeal pituitary) to the base of the brain. The pituitary stalk is the main neural connection between the hypothalamus (median eminence) and the posterior lobe of the pituitary. In addition to these neurons, there are the capillary loops of the hypothalamic-pituitary portal system providing the principal blood supply to the various portions of the anterior lobe.
Pituitary stalk interruption syndrome (hypoplasia of the anterior lobe, ectopic position of the posterior lobe, and interruption of the stalk) is strongly correlated with multiple anterior pituitary hormone deficiencies.6 Injury may be developmental or secondary to transection of the stalk. A number of other anomalies of the craniofacial area may coexist with hypopituitarism. The syndrome of basal encephalocele and hypothalamic–pituitary dysfunction should be mentioned because these patients all have a nasoepipharyngeal mass or unexplained “nasal polyp.”7 The diagnosis should be considered when evaluating a nasal mass, especially in conjunction with the associated findings of hypertelorism, broad nasal root, midline facial defects, optic atrophy, or optic coloboma.
Inherited Pituitary Disorders
GENETIC DISORDERS OF GROWTH HORMONE DEFICIENCY. Five to 30 percent of children with growth hormone deficiency have an affected first-degree relative, which is consistent with a genetic cause. These genetic anomalies may result in isolated or combined pituitary hormone deficiencies. The more common are POU1F1, homolog of the mouse Pit-1, and PROP-1 deficiencies, which lack not only GH, prolactin (Prl), and thyroid-stimulating hormone (TSH), but also the gonadotropins. The specific genes
encode members of the homeodomain family of transcription factors, which play an important role in the development of the human pituitary gland.8 Other deficiencies are caused by growth hormone-releasing hormone receptor mutations.
encode members of the homeodomain family of transcription factors, which play an important role in the development of the human pituitary gland.8 Other deficiencies are caused by growth hormone-releasing hormone receptor mutations.
ISOLATED GROWTH HORMONE DEFICIENCY AND PITUITARY DWARFISM. The classification of familial GH deficiency primarily is descriptive and is based on the inheritance of the appropriate phenotype and lack of response to pharmacologic stimuli for GH secretion.9 Six distinct groups based on the mode of inheritance and other hormone deficiencies have been defined (see Table 18-2). In only one, isolated GH deficiency type IA, have the pathophysiologic characteristics been defined: absence of the structural gene for GH (hGH-N).10 The others apparently are a result of the lack of synthesis or secretion of the hypothalamic-releasing hormone, growth hormone–releasing hormone (GHRH), or to an excessive secretion of the inhibitory hormone, somatostatin. However, heterogeneity of structure and function exists within and among families with isolated GH deficiency and within and among families with pituitary deficiency, making the determination of the precise location of the defect difficult—hypothalamus or pituitary.11
Boys with severe GH deficiency and other anterior pituitary deficits may present with microphallus and hypoglycemia. The physical examination of the genitalia should alert the physician to determine the level of plasma glucose frequently and to use GH and cortisol to treat refractory hypoglycemia.12
Congenital Tumors
CRANIOPHARYNGIOMA. The most common tumor in the area of the pituitary of children is the craniopharyngioma13 (see Chap. 11). It arises from the embryonic remains of the craniopharyngeal duct (Rathke pouch) and is of epithelial origin. This neoplasm commonly is cystic and often contains dark, thick, viscous fluid (“machinery oil”). Although congenital, it is so slow-growing that signs and symptoms often are not manifested until late in the first decade or in the second decade of life or even into adulthood. In addition to the signs of hypopituitarism, children and adolescents may have neurologic symptoms, including prolonged frontal headache, vomiting, or vision deficits—decreased acuity, diplopia, or photophobia. On examination, papilledema, optic nerve atrophy, and impaired visual fields are found in most; they indicate raised intracranial pressure. Older children and adolescents may have obesity or amenorrhea. In the asymptomatic patient, the diagnosis may be made by noting flecks of calcium in the suprasellar region or changes within the suprasellar area as evaluated on computed tomographic (CT) or magnetic resonance imaging (MRI; Fig. 18-4) studies. In a symptomatic patient, CT or MRI scanning is extremely helpful in confirming the diagnosis and determining the extent of the neoplasm.
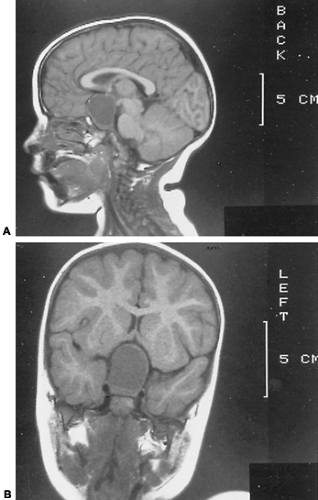 FIGURE 18-4. Computed tomographic (CT) scans of the brain of a 3-year-old girl with a craniopharyngioma. A, Sagittal view showing large suprasellar mass. B, Coronal view showing compression of pituitary gland and large suprasellar mass. (Courtesy of Dr. W. Cail, University of Virginia.) |
Although craniopharyngioma is most commonly found in children, optic and third ventricle gliomas and arteriovenous malformations must be considered. The treatment is surgical
and may be possible through the transsphenoidal approach. Aspiration of the cyst can decompress the mass and relieve symptoms but rarely is curative. Radiation therapy including intracystic application of radioactive pellets, although controversial, may be effective as adjuvant therapy to surgery. Tumor recurrence, even after “total” removal, is common. Hypopituitarism, especially GH deficiency, may be found preoperatively. Postoperatively, it is present in most patients, often in association with other defects in anterior pituitary function and central diabetes insipidus. Meticulous follow-up of growth velocity and pituitary target-organ function, and early replacement therapy greatly decrease the morbidity and mortality after surgery.
and may be possible through the transsphenoidal approach. Aspiration of the cyst can decompress the mass and relieve symptoms but rarely is curative. Radiation therapy including intracystic application of radioactive pellets, although controversial, may be effective as adjuvant therapy to surgery. Tumor recurrence, even after “total” removal, is common. Hypopituitarism, especially GH deficiency, may be found preoperatively. Postoperatively, it is present in most patients, often in association with other defects in anterior pituitary function and central diabetes insipidus. Meticulous follow-up of growth velocity and pituitary target-organ function, and early replacement therapy greatly decrease the morbidity and mortality after surgery.
DIENCEPHALIC SYNDROME. The diencephalic syndrome usually comprises a tumor in the diencephalon and the clinical picture of severe emaciation with relative conservation of growth rate, alert appearance, and relatively few neurologic signs. It is nearly always, but not exclusively, found in infancy.14,15 There is a striking lack of subcutaneous fat. Often, the child seems inappropriately happy (Fig. 18-5). Some clinicians believe that it is related to the age of onset of compression of the hypothalamus because it is not seen in patients with craniopharyngiomas or other tumors within the same anatomic area that present later in childhood, although such tumors displace the third ventricle in a manner similar to that of opticochiasmatic gliomas. Although endocrinologic deficits may be present, they are inconstant and do not help in the diagnostic process. Most tumors are gliomas located in the anterior hypothalamus or in the optico-chiasmatic system. The syndrome in infants with tumors placed more posteriorly is characterized by the early onset of vomiting and the absence or late onset of nystagmus, tremor, pallor, polyuria, papilledema, or optic atrophy. These patients are more likely to have malignant cells and raised protein concentrations in the cerebrospinal fluid than are children with more anteriorly placed tumors. These latter patients more often have nystagmus and optic atrophy. Vomiting appears later.16
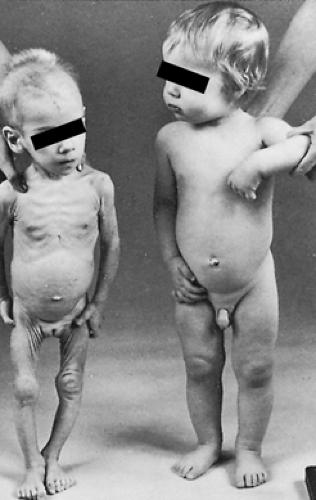 FIGURE 18-5. A 10-month-old child with diencephalic syndrome (left next to a normal child of the same age (right). Despite striking emacia tion, the infant appeared happy and alert and was extremely hyperac tive. There was nystagmus. The feet and hands appeared large in comparison with the body. Basal growth hormone levels were high and were not suppressed adequately with glucose administration. At cra niotomy, there was an optic glioma involving the optic nerves and chi asma that extended into the anterior hypothalamus and the posterior portion of the third ventricle. (From Häger A, Thorell JI. Studies on growth hormone secretion in a patient with the diencephalic syndrome of emaciation. Acta Paediatr Scand 1973; 62:231.) |
Radiographic studies in patients with anteriorly placed tumors may show enlarged optic foramina and, less commonly, sellar alterations, evidence of increased intracranial pressure, and, rarely, calcification. Computed tomographic and MRI scans may be the best tools for defining the exact nature and extent of disease because the optic nerves, chiasm, and sellar and suprasellar regions can be visualized precisely.
The prognosis is poor, and the treatment remains controversial. Surgery is rarely curative, but it is important to obtain a pathologic diagnosis. Radiation therapy can dramatically reduce the mass of the tumor. However, the natural history is uncertain and variable. Thus, treatment for these often slow-growing tumors varies from no therapy, to surgical excision, to radiation therapy; however, one must be concerned with ultimate brain growth if a child younger than 2 to 3 years old undergoes radiation therapy. Because most series have been small and their data were collected over several decades when the approach to diagnosis, therapy, intensive postoperative care, and endocrinologic replacement therapy was in great flux, it is not surprising that no single therapeutic protocol has proved to be clearly superior.
Defects in the Structure, Metabolism, or Secretion of Growth Hormone or Insulin-Like Growth Factor I
BIOLOGICALLY INACTIVE GROWTH HORMONE. Children with growth failure who have normal or elevated basal GH concentrations or a normal or increased response to pharmacologic stimuli, but diminished IGF-I concentrations that cannot be attributed to malnutrition, chronic illness, or other causes, may secrete a bioinactive (subactive) GH molecule. Bone and dental development are significantly delayed, and the body proportions are more appropriate for chronologic than height age. Although some investigators have proposed this hypothesis for short stature in the few children who fit into the diagnostic category, there is sparse evidence. Clearly, it is not the public health problem it was originally considered.17 Findings in a single patient indicate that aggregation of serum GH may be responsible.18 However, a mutation in the GH gene itself can produce a mutated form of GH (found to be expressed in Escherichia coli), which is clinically associated with short stature in the affected child.19 The mutated GH binds avidly to the GHG receptor in the IM-9 cell line, but does not stimulate intracellular signaling pathways. Thus, it inhibits the bioeffects of native GH.
The growth hormone insensitive syndrome (GHIS) represents another defect in the mechanism of GH action3,20 (Fig. 18-6). These patients with familial dwarfism and clinical features of GH deficiency tend to be of normal birth weight, but growth velocity is retarded soon after birth. Motor development, bone maturation, and dental eruption are slow, and the anterior fontanelle may close later than average. The facial bones grow more slowly than the cranial vault. When the child has a small mid-face and mandible and a bulging forehead, macrocephaly should be considered.
The children tend to be obese and have a high-pitched voice. Although many of the original patients were of Semitic origin, subsequent patients have come from many ethnic backgrounds.
The children tend to be obese and have a high-pitched voice. Although many of the original patients were of Semitic origin, subsequent patients have come from many ethnic backgrounds.
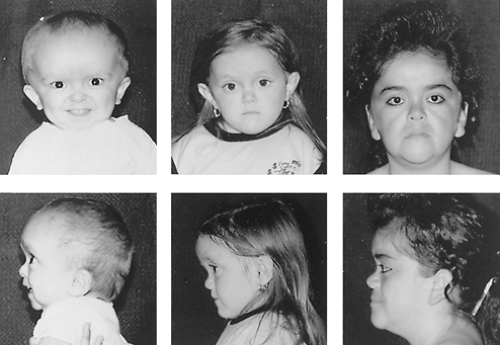 FIGURE 18-6. Three Ecuadorian patients with growth hormone receptor deficiency resulting from a point mutation at codon 180 of exon 6 of the growth hormone receptor: a boy aged 26 months (height, -8.2 SD), a girl aged 4.8 years (height -7.4 SD), and a woman aged 19 years (height, -6.5 SD). The vertical dimension of the face is decreased, the nasal bridge is hypoplastic, and the forehead is of normal dimension, giving the impression of craniomegaly, especially in the children. (Courtesy of J. Guevara-Aguirre, Institute for Endocrinology, Metabolism, and Reproduction, Quito, Ecuador; and A.L. Rosenbloom, University of Florida College of Medicine.) |
GH values are high basally and are elevated in response to pharmacologic stimuli, but IGF-I levels are low. The circulating GH molecules are biologically active. The pathogenesis of the defect is failure of the liver to respond to GH by generating IGF-I. Because the extracellular domain of the GH receptor is a growth hormone–binding protein (GHBP), this protein might be expected to be absent or defective in GHIS.21 The initial direct evidence for a GH receptor defect in GHIS was that hepatocytes obtained at biopsy from a patient with this disorder did not bind tracer quantities of radiolabeled GH, although samples from control subjects undergoing abdominal surgery did.22 Subsequently, the similarity of the circulating GHBP to the GH receptor (the former is the extracellular domain of the latter) led to the finding that GHBP was absent in patients with GHIS. The clinical and biochemical characteristics of GHIS have been reviewed.23 Prominent biochemical abnormalities include low circulating levels of IGF-I, IGF-II, insulin-like growth factor–binding protein-3 (IGFBP-3), and GHBP; and levels of IGF-I, IGF-II, and IGFBP-3 that are higher in adults than in children without a change in the level of GHBP.22 A number of treatment trials with recombinant human IGF-I are under way. In all the young patients, the growth rate is accelerated. Many patients had growth rates during therapy in the range of 8 to 10 cm per year. Because of the low IGFBP-3 levels at the onset of treatment and the lack of a buffer for the injected IGF-I, some patients experienced hypoglycemia.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


