DIABETES INSIPIDUS
ETIOLOGY
In theory, any of a series of defects in the vasopressin neurosecretory process can be implicated as the cause of hypothalamic diabetes insipidus.3 Abnormalities may arise in the osmoreceptor that controls vasopressin secretion, even when the thirst osmoreceptor is spared. Alternatively, abnormalities may involve the synthesis and packaging of vasopressin (including genetic defects), damage to the vasopressinergic neurons, or disorders of neurohypophyseal hormone release. Enhanced inactivation of vasopressin by circulating degrading enzymes or antibodies is another potential cause of decreased antidiuretic activity.
In practice, however, most cases of permanent hypothalamic diabetes insipidus are caused by damage to the hypothalamo-neurohypophyseal area. The most common causes of this condition are listed in Table 26-1.4
 |
FAMILIAL ABNORMALITIES
Studies have revealed exciting data regarding a variety of genetic abnormalities found on chromosome 20 in several kindreds with autosomal dominant hypothalamic diabetes insipidus. The first report on different families showed single nucleotide substitutions in the region coding for neurophysin (glycine to serine at position 57).5 This mutation is presumed to interfere with the normal vasopressin-neurophysin tetramer complex formation that occurs in the packaging and transport of vasopressin to the neurohypophysis. Two groups investigating another extended family discovered a nucleotide substitution in the signal peptide (alanine to threonine at position -1).6,7 The signal peptide directs the prohormone to the endoplasmic reticulum, where it is cleaved. Both groups speculate that the mutation alters the cleavage mechanism, resulting in abnormal processing of the prohormone. After the description of the first genetic abnormalities causing familial hypothalamic diabetes insipidus, more than 22 different kindreds with unique genetic mistakes have been documented8 (Fig. 26-1).
TRAUMA
Closed head trauma or frank damage to the pituitary stalk or hypothalamus as a result of surgical intervention is often the cause of a form of diabetes insipidus that usually presents within 24 hours of injury. In ˜50% of cases of post-traumatic diabetes insipidus, the condition resolves spontaneously within a few days. Permanent diabetes insipidus develops in another 30% to 40% of these patients, and the remainder exhibit a triphasic response to injury. In the last group, the onset of polyuria is abrupt and the condition lasts a few days. It is followed by a period of antidiuresis that may last 2 to 14 days before permanent diabetes insipidus develops. This triple response to injury is believed to be attributable to release of the vasopressin that is stored in granules.11 Recognition of this entity by clinicians
should help to prevent inappropriate treatment that would result in hyponatremia during the second of the three phases.
should help to prevent inappropriate treatment that would result in hyponatremia during the second of the three phases.
TUMORS
Tumors of the anterior pituitary rarely cause diabetes insipidus. In a series of >100 cases of hypothalamic diabetes insipidus, 13% were attributable to tumors, which included glioma, germinoma, and craniopharyngioma.12 In children, a central tumor is a frequent cause of hypothalamic diabetes insipidus, accounting for ˜25% of cases; in this population, the most common intracranial tumor is germinoma.13 Metastatic deposits in the hypothalamus causing diabetes insipidus usually arise from carcinoma of the breast or bronchus.
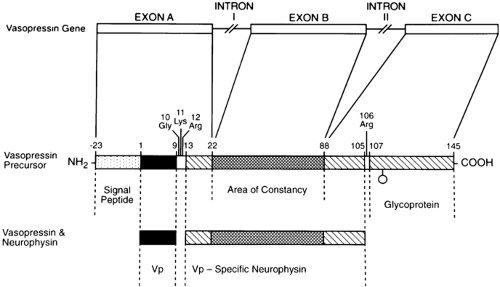 FIGURE 26-1. Vasopressin gene, vasopressin precursor molecule, and vasopressin with its specific neurophysin. Three exons encode for the precursor molecule, which comprises a signal protein, vasopressin hormone, neurophysin, and a glycoprotein moiety coupled by amino acids. Mutations in the vasopressin gene have been located in all parts of the precursor molecule except vasopressin itself. (Vp, vasopressin.) |
GRANULOMATOUS DISEASE
Granulomatous disease accounts for only a few cases of diabetes insipidus in adults (i.e., sarcoidosis, tuberculosis). However, in children with granulomatous disease, histiocytosis X may cause as many as 40% of pediatric cases.
IDIOPATHIC
Idiopathic hypothalamic diabetes accounts for ˜25% of all cases.12 One-third of patients with apparent idiopathic disease have circulating antibodies to the vasopressin-producing cells in the hypothalamus, a finding which suggests an autoimmune origin for the disorder.14 Some patients have an acute lymphocytic infiltration of the infundibulum and neurohypophysis that can be demonstrated on open biopsy and subsequently resolves.15
NEPHROGENIC DIABETES INSIPIDUS
Mild forms of nephrogenic diabetes insipidus are relatively common (see Table 26-1). Mechanisms responsible for renal resistance to the antidiuretic effect of vasopressin may occur at one or more of the many different sites in the chain of biochemical responses to vasopressin.11,15a Chronic renal disease secondary to numerous conditions, many drugs (e.g., lithium15b), prolonged electrolyte disturbances from hypokalemia, and hypercalcemia account for most cases of nephrogenic diabetes insipidus. The inherited forms of nephrogenic diabetes insipidus are rare, and cause severe polyuria, dehydration, and failure to thrive in the young. With the identification of the V2 (antidiuretic) receptor gene on the X chromosome, a variety of substitutions, mutations, or premature stops have been isolated in kindreds with this disorder that cause defects in the trans-membrane V2 receptor.16 Studies involving three other families with congenital nephrogenic diabetes insipidus have shown autosomal inheritance of the disorder, in contrast to the more common X-linked form due to genetic mutations of the V2 receptor gene localized to the Xq28 region of the long arm of the X chromosome. The autosomal form is due to novel genetic mutations of the gene encoded for the vasopressin-sensitive water channel protein, aquaporin-2, which is located in the collecting tubules.17
DIPSOGENIC DIABETES INSIPIDUS
Dipsogenic diabetes insipidus, also called primary polydipsia or habitual water drinking, is often psychogenic in origin. The course of polyuria in psychotic patients is variable, with fluctuations in polydipsia and urine volumes occurring over the years. Occasional patients with hypothalamic diabetes insipidus who are treated with antidiuretic preparations continue to have polydipsia and, consequently, run the risk of developing hyponatremia. Whether these patients continue to drink because of habit or because of a hypothalamic lesion affecting the thirst osmoreceptor is unknown. A few structural abnormalities resulting in increased thirst have been reported. Drugs that cause dryness of the mouth (e.g., thioridazine hydrochloride) may increase drinking but do not result in true polydipsia, in contrast to lithium, which can stimulate thirst directly.
CLINICAL FEATURES
In adults, the major clinical manifestations of diabetes insipidus include the frequent passage of large volumes of dilute urine (often both day and night), excessive thirst, and increased fluid ingestion. Patients with mild degrees of diabetes insipidus may consider their symptoms to be so minimal that they fail to seek medical attention. However, the severity of diabetes insipidus varies widely, with 24-hour urine volumes ranging from 2.5 to 20 L. Even with the most extreme forms of the disorder, patients maintain their water balance as long as thirst appreciation remains intact and adequate volumes of fluid are ingested.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


