PATHOPHYSIOLOGY
SITES OF NONTHYROIDAL ILLNESS INTERACTION WITH THYROID ECONOMY
Every accessible point in thyroid hormone homeostasis has been examined or considered as a potential contributor to the thyroidal effects of NTI (Fig. 36-3). A number of studies have examined the role of cytokines in the mediation of these effects (discussed later). At the hypothalamic-pituitary level, decreases in mRNA for TRH have been noted in the hypothalamus obtained at autopsy from patients succumbing to an NTI.43 In these patients, TRH gene expression was inversely correlated with antemortem serum T3 levels. Other endogenous modulators of the hypothalamic–pituitary–thyroid axis that potentially are activated during NTI include cortisol, somatostatin, dopamine, and the β-endorphins. Within the pituitary, several potential
defects, including altered tissue levels of T444 or of the T3 receptor45; enhanced activity of the type II 5′-monodeiodinase enzyme13; and diminished synthesis,46 release,19 or bio-activity47,48 of TSH, have been considered as possible explanations for the apparently blunted pituitary responsiveness that occurs in NTI. Multiple disturbances, rather than any single defect, probably account for the diverse changes in the hypotha-lamic-pituitary control of thyroid function observed in NTI.
defects, including altered tissue levels of T444 or of the T3 receptor45; enhanced activity of the type II 5′-monodeiodinase enzyme13; and diminished synthesis,46 release,19 or bio-activity47,48 of TSH, have been considered as possible explanations for the apparently blunted pituitary responsiveness that occurs in NTI. Multiple disturbances, rather than any single defect, probably account for the diverse changes in the hypotha-lamic-pituitary control of thyroid function observed in NTI.
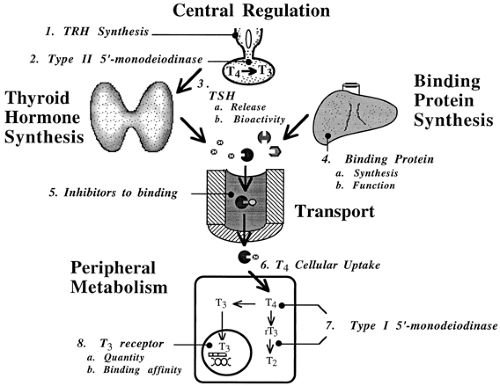 FIGURE 36-3. Potential sites of interaction between thyroid economy and nonthyroidal illness. Interaction at multiple levels of regulation has been demonstrated or hypothesized (see text). (TRH, thyrotropin-releasing hormone; T4, thyroxine; T3, triiodothyronine; TSH, thyroid-stimulating hormone; rT3, reverse T3; T2, diiodothyronine.) |
Much has been learned about thyroid hormone production, binding defects, and peripheral metabolism in NTI. Decrements in circulating thyroid hormone levels are well-established features of NTI, yet the production and release of serum T4 is normal in these disorders. Furthermore, a decreased production or an accelerated loss (nephrotic syndrome) of binding proteins accounts for only a small portion of the observed decreases in serum T4 and T3 levels. Circulating inhibitors of thyroid hormone binding have been postulated to contribute to the lowering of serum total T4 levels12 and the decreased T4/TBG ratio49 that occurs in NTI in the face of normal or high serum FT4 concentrations. Free fatty acids have been investigated in this regard.10 However, the nature and even the existence of these inhibitors remains controversial, and alternative explanations have been proposed for the binding abnormality in NTI.50 Defects in TBG itself, as evidenced by relative increases in an isoform that has an altered electrophoretic motility, have been reported to affect thyroid hormone levels in NTI.51 Altered metabolism of T4 resulting from decreased activity of type I 5′-monodeiodinase activity is the best known, yet still incompletely understood, phenomenon noted in NTI. The result of this enzymatic inhibition is the striking decrease in serum T3 and elevation in rT3 that is characteristic of the euthyroid sick syndrome. Diminished hepatic uptake of T4 may also contribute to decreased peripheral production of T3 in patients with NTI.52 Interestingly, rT3 may compete with T4 as a substrate for both type I and type II 5′-monodeiodinases, compounding the defect in T4-to-T3 conversion.11 Other nonpharmacologic mechanisms contributing to the inhibition of 5′-monodeiodination that occurs during NTI remain to be elucidated. The cloning of the complementary DNA for human type I 5′-monodeiodinase53 has allowed the demonstration of decreased expression of this gene in an animal model of NTI.54
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


