Graft-Versus-Host Disease and Graft-Versus-Tumor Response
Graft-Versus-Host Disease and Graft-Versus-Tumor Response
Joseph Pidala
Frederick L. Locke
Claudio Anasetti
INTRODUCTION
Graft-versus-host disease (GVHD) is the most common treatment complication following hematopoietic stem cell transplantation (HSCT) from an allogeneic donor, and it is a major cause of recipient morbidity and mortality. GVHD results from the recognition of recipient tissue antigens by immune competent T cells transplanted with the graft. There is an acute form of GVHD with a rapid onset, usually early after transplantation, and a chronic form of GVHD with late onset. These two GVHD syndromes are largely distinct in pathogenesis, clinical manifestations, prevention, and treatment, and therefore are presented separately in this chapter. Donor cells in the graft also produce an immune response against targets in the recipient malignant cells, through a reaction that contributes substantially to the antitumor activity of allogeneic HSCT. Such a graft-versus-tumor (GVT) response is presented in the third section of this chapter.
ACUTE GRAFT-VERSUS-HOST DISEASE
Pathophysiology
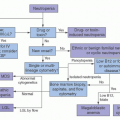
The requisites for the development of GVHD were recognized over 50 years ago by Billingham: (1) the inclusion of immune competent cells in the graft, (2) the inability of the recipient to reject the graft, and (3) the presence of recipient tissue antigens foreign to the donor. Animal model studies, primarily in rodents, led to a three-step model of GVHD pathophysiology that simplifies the complex immune interactions by placing emphasis on cellular interactions and cytokines: (1) host antigen-presenting cell (APC) activation, (2) donor T-cell activation, and (3) pathogenic effector cells and inflammatory mediators produce the disease.1, 2 GVHD biology is extremely complex involving intracellular signaling, soluble mediators, and cellular interactions. Positive and negative signals exist at multiple levels including the cell surface, signaling protein networks, transcription promoters, mRNA, and posttranslational modification. In addition to histocompatibility antigen disparities between donor and recipients, genetic polymorphisms at any of these immune checkpoints may promote or prevent GVHD. The interaction of immune cells, visceral organ tissue, cytokine, and chemokine signals, cellular trafficking into and out of lymphoid and visceral organs is similarly complex. Recent attempts to rein in these numerous, continuous, and interrelated processes led Paczesny and Reddy to create a comprehensive model of GVHD pathophysiology based on the framework of normal immune system interactions. It is the expected behavior of donor immune cells to cause pathophysiology within the host that harbors unrecognized antigens which are treated as foreign by the donor cells. Put more simply, acute GVHD arises when donor T cells react in a physiologically appropriate way against recipient proteins recognized as foreign. However, the signaling events, intracellular pathways, and immune cell trafficking are extremely complex and remain to a large degree incompletely understood. This proposed nonlinear model consists of four interrelated elements to describe the role of immune system recognition and activation at the genetic, cellular, and cytokine level: triggers, sensors, mediators, and effectors of GVHD (Table 105.1).3
Critical to acute GVHD onset is the degree of genetic variability between donor and host, the level and site of tissue damage set forth by the conditioning regimen, the ability of host or donor cells to present antigen, the cellular and cytokine mediators of response, and the nature and degree of the immune effector activity.
Triggers
Human leukocyte antigens (HLAs) are encoded by the major histocompatibility gene complex (MHC) on chromosome 6p and function as major histocompatibility antigens in transplantation. The natural function of HLA is to present antigenic peptides to specific T cells. Exogenous and endogenous proteins present within human cells are continually broken down into peptides. HLA molecules on professional APCs bind those peptides within their groove and present them from the cell surface to the T-cell receptors on T lymphocytes. Inasmuch as the human genome includes greater than 107 polymorphic sequences, there is great likelihood that donor and recipient differ for one or more of the proteins that are presented as HLA:peptide complexes to T cells. These polymorphic proteins function as minor histocompatibility antigens (mHAs) in transplantation. Autoreactive T-cell recognition of “self” peptides in the thymus leads to negative selection (death), so that the probability of an individual developing autoimmunity is minimized. After transplantation, this delicate balance of self and nonself is perturbed. Donor and recipient may differ for HLA molecules or mHA. Each peripheral T cell harbors a unique T-cell antigen receptor; therefore, the human repertoire for recognition of foreign antigens is enormous. Donor and recipient HLA mismatching plays a critical role in the development of GVHD, because each type of HLA presents a unique repertoire of antigenic peptides. When recipient cells are mismatched at an HLA locus, the donor T cells have not undergone thymic deletion to avoid recognizing those HLA:peptide complexes. Therefore, the degree of HLA disparity directly increases the probability and severity of acute GVHD following transplantation.4, 5 For umbilical cord blood donors a greater degree of HLA mismatch is tolerated presumably due to the small number of T cells within the cord blood (˜104 per kg recipient weight) as compared to adult donor blood (˜108) or marrow (˜107).
mHA are polymorphic proteins genetically encoded anywhere outside HLA. Host mHA proteins are presented as HLA:peptide complexes to donor T cells and elicit an immune response even when presented via a matched HLA molecule. mHA mismatches are only partially characterized for the ability to affect acute GVHD. Some mHA have broad tissue expression such as the male-associated H-Y antigen.6 Alternatively, HA-1 and HA-2, for example, have their expression restricted to hematopoietic cells, including leukemia, and immune cells.7 It is this variability of tissue mHA expression that may allow for tailoring therapy toward hematopoietic-restricted mHA to promote GVT response without GVHD. Efforts to prevent GVHD would have to target immune responses against the ubiquitously expressed mHA.6, 8, 9
Other triggers of GVHD include molecules that activate the innate immune system. Pre-transplant radiation or chemotherapy damages host cells and enteric microbes that contain damage-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs), respectively. DAMPs such as proteases or ions released from damaged epithelial cells and PAMPs such as lipopolysaccharide within common bacteria, can activate Toll-like receptors setting off a cascade of immune events characterized by proinflammatory cytokine secretion.10 To a large degree these processes are responsible for initiation of the “cytokine storm” described as critical in earlier models of acute GVHD.11
Mismatches in HLA molecules from donor to host lead to recognition of the cell as foreign by T cells.
mHA
Normal variability of protein within genome
mHA are recognized as foreign by MHC:T-cell interactions.
DAMPs and PAMPs
Common organic patterns originating from damaged cells or pathogens triggering innate immunity
DAMP release after cell death due to conditioning therapy or PAMP presence via pathogens triggers innate immunity and cytokine cascade.
Sensors
Antigen presenting cells
Present antigens to effectors
Present exogenous and endogenous protein sequences to T cells via direct, indirect, or cross presentation.
Mediators
CD4 T helper subsets
Skew effector responses to the appropriate degree
Effects of T-cell subsets on graft-versus-host disease incompletely understood.
Regulatory T cells
T-cell subset which acts to prevent untoward autoimmunity
Donor regulatory T cells play a role in reducing graft-versus-host disease.
Inhibitory signals
Signals provided in concert with antigen presentation to decrease the immune response
Co-stimulatory signals
Signals provided by professional antigen presenting cells to initiate immune response
Effectors
Activated CD4 and CD8 T cells
Lead to killing or neutralization of foreign cells or debris via IFNγ, perforin, or granzyme secretion
Donor cells act to damage host cells and organs leading to manifestations of graft-versus-host disease.
HLA, human leukocyte antigen; DAMPs, damage-associated molecular patterns; mHA, minor histocompatibility antigen; MHC, major histocompatibility complex; PAMPs, pathogen-associated molecular patterns.
Adapted from Paczesny S, Hanauer D, Sun Y, Reddy P. New perspectives on the biology of acute GVHD. Bone Marrow Transplant 2010;45:1-11.
Sensors
Sensors of GVHD refer to the cells and processes that recognize the mHA or HLA mismatches as foreign, thereby setting the immune response in motion. Antigens are presented by host professional APC such as dendritic cells, macrophages, or Langerhans cells. Host dendritic cells may be primed for antigen presentation via the cytokine storm accompanying the conditioning therapy. The nature of antigen presentation interactions includes direct presentation, which mimics physiologic APC-T-cell interactions. Specifically, direct presentation consists of host APC presenting endogenous (cytosolic) host peptide to donor CD8 T cells, and exogenous (extracellular, endocytosed peptide, processed in lysosomes) antigens are presented to donor CD4 T-cells. Indirect presentation specifies exogenous antigen presented from donor APC to donor CD4 cells. Cross-presentation refers to exogenous antigen (derived from host cells) presented to donor T cells from donor APC. Over time APC change from primarily host origin to donor origin. It is likely that direct presentation by host APC is predominant during early stages of acute GVHD, whereas indirect or cross-presentation by donor APC is predominant in chronic GVHD. APC-T-cell interaction involves not only interaction between HLA plus cognate antigen with T cell receptor (TCR), but co-stimulatory and inhibitory signals at the immune synapse, secondary signals from cell subsets, and paracrine or autocrine mediators. APC provide necessary co-stimulatory signals directly when activating T cells, such as CD28, CD40, and ICOS as well as inhibitory signals such as CTLA-4 and PD-1. Manipulation of these signals via antibody interference or small molecule blockade of downstream signaling events remains a field ripe for modulation of GVHD. Exogenous cell types critical to inhibition of APC include regulatory T cells, gamma-delta T cells in the GI tract, natural killer (NK) cells, and host NKT cells. T-cell response is an adaptive immune response which is often triggered by the innate immune response. As described, Toll-like receptors are sensors of the DAMP and PAMP release precipitated by the condition regimen. These Toll-like receptor interactions lead to release of proinflammatory cytokines such as tumor necrosis factor alpha (TNFα) and interleukin (IL)-6 which contribute to the inflammatory milieu surrounding T-cell-mediated GVHD damage.
Mediators
GVHD is primarily mediated by donor T cells, and differing subsets that have normal physiologic activities outside the framework of transplant, act at different capacities to induce or inhibit GVHD development. Most animal models point to the relevance of naive T cells, rather than memory T cells, in the induction of GVHD. The process of homeostatic proliferation in the lymphoablated host causes brisk clonal expansion of the transferred donor T cells and plays an important role for the initiation of acute GVHD. Donor regulatory T cells (Treg) repress the GVHD responses. Treg are CD4 T cells that express the FOXP3 transcription factor, high levels of the alpha chain of the IL-2 receptor, but no IL-7 receptor and function as a natural suppressor to maintain peripheral tolerance. These donor-derived regulatory T cells inhibit the activation and proliferation of donor T cells implicated in the pathogenesis of acute GVHD and may spare the GVT effect.12 Recent data show that low-dose IL-2 expands regulatory T cells in vivo and may be effective therapy for established GVHD,13 whereas anti-IL-2 receptor antibodies, tested before Treg importance and biology were recognized, appear to accelerate GVHD.14
Additional CD4 T-cell subsets are critical. It is Th1 cells, characterized by IL-2 and interferon gamma (IFNγ) secretion, which are the likely main T-cell mediators of acute GVHD. Th2 T cells have been linked to protection against acute GVHD. Th17 CD4 cells secrete IL-17 and function distinctly from Th1 cells in a manner that is not yet fully characterized in the context of human GVHD. Murine transplant models provide evidence that Th17 can cause GVHD.15, 16 T-cell trafficking is another important step in GVHD pathogenesis. Differential expression of chemokines on GVHD target organs and draining lymph nodes mediate differential trafficking of T-cell subsets expressing different chemokine receptors. An initial report indicated that a CCR5 inhibitor can prevent GVHD in gut and liver.17
Effectors
The effectors of acute GVHD are the cytolytic cellular elements and inflammatory cytokines. Activated CD4 and CD8 cytotoxic T lymphocytes are the primary effectors of GVHD. Upon interaction with their cognate HLA:peptide they act to lyse or cause apoptosis of target cells via secretion of perforin and granzyme or binding of FAS, respectively. IFNγ and TNFα are both critical to the inflammatory process of GVHD. Inflammatory cytokines induce and amplify cellular damage.
EPIDEMIOLOGY
The major source of treatment-related morbidity and mortality is GVHD. The incidence of clinically relevant (grade II to IV) acute GVHD ranges from 35% to 80%, with a higher incidence reported for recipients of unrelated donors compared to matched related donors.18, 19 The day 100 incidence of severe acute GVHD (grades III-IV) occurs in approximately 15%, but may be as high as 35% depending upon risk factors such as HLA disparity.5
RISK FACTORS
Risk factors for the development of acute GVHD include T-replete transplant, recipient and donor HLA disparity, female donor for a male recipient, donor and recipient age, hematopoietic stem cell source (peripheral blood progenitor cells [PBPCs] > marrow > cord blood), graft cellular composition (worse with higher T-cell and CD34 cell numbers), higher conditioning intensity, diagnosis (worse with chronic myeloid leukemia), and immune response gene polymorphisms.20, 21 Predictive factors are of great interest to stratify patients prior to development of serious morbidity or mortality and to focus prevention upon the proper biological parameters.
HLA matching is critical to prevent acute GVHD. A single allele mismatch at HLA-A, HLA-B, HLA-C, or HLA-DRB1 increases the likelihood of acute GVHD development, and mismatch for multiple alleles compound the risk.5, 22
Female donor for a male recipient and donor parity are risk factors for the development of acute GVHD. The increased risk for male recipients of female as opposed to male donors is attributed to the recognition of H-Y mHAs by female donor T cells.23, 24 During pregnancy, female donors develop an immune response to the paternal mHAs of the fetus and mount a secondary, augmented T-cell alloimmune response against the same mHAs if expressed in the recipient.25
Increasing donor age is associated with increased risk for the development of severe acute and chronic GVHD, and worse mortality.26 Age of the recipient is also important with higher rates of acute GVHD in older compared to younger cohorts.20, 27
The rate of chronic GVHD in patients receiving PBPC grafts is higher compared to bone marrow; however, the association of PBPC with acute GVHD is not clearly established.28, 32 Unrelated cord blood transplant is associated with a low rate of acute GVHD when compared to transplant with similarly HLA mismatched adult donors.33, 34 Multiple factors may contribute to this effect; however, the low number of T cells in the graft and T-cell naive likely play a protective role.
The conditioning regimen used affects the incidence of acute GVHD. With high-dose intensity conditioning the incidence of acute GVHD is higher. For example, increasing doses of radiation can double the incidence of acute GVHD.35 Several large retrospective studies showed that reduced intensity regimens lead to reduced rates of grade II-IV acute GVHD when compared to higher intensity regimens.36, 37 Animal models suggest the correlation of the incidence of GVHD with intensity may be due to increased damage to host tissue and amplified release of cytokines followed by activation of APC.38 In addition, higher intensity regimens decrease the fraction of recipient T cells that persist after the regimen, leading to a lower barrier to donor engraftment and greater homeostatic re-population by donor T cells, which is associated with worse acute GVHD.39, 40
The cellular composition of the graft is linked to the incidence of GVHD. Despite the link between chronic GVHD and CD34 cell dose, conflicting studies outline the importance of CD34 cell dose on acute GVHD.41, 42 The preponderance of evidence suggests that there is no correlation between CD34 cell dose and the incidence of acute GVHD, whether the progenitor source is marrow or PBPC,43, 44, 45 although one study, looking at patients given cyclosporine as a prophylactic agent, revealed a positive association of CD34 dose with acute GVHD.46
Genetic polymorphisms of immune response genes are associated with an increased incidence of acute GVHD. Single nucleotide polymorphisms (SNPs) can alter cytokine binding domains, thereby altering affinity in a functional or nonfunctional way. Particular SNPs in genes coding IL-10, IL-6, IL-2, HPSE, CTLA-4, and MTHFR have all been identified as increasing the risk of clinically relevant or severe acute GVHD, likely via increased level or activity of these cytokines or receptors.47
Cytokine biomarkers correlate with the presence of acute GVHD and provide prognostic information independent of the severity of GVHD. Although not widely in diagnostic use, the panel which includes IL-2-receptor-alpha, TNF-receptor-1, IL-8, and hepatocyte growth factor, can confirm the diagnosis at the onset of clinical symptoms.48 An expanded 6 biomarker panel which also includes elafin, a skin-specific marker, and regenerating islet-derived 3-α, a gastrointestinal (GI) tract-specific marker was shown to predict response rate at day 28 after onset and survival at day 180 after onset, for patients with acute GVHD. This panel was predictive at 3 time points; onset, 2 weeks, and 4 weeks into therapy. Future clinical trials will determine if such stratification into high and low risks groups will lead to improved outcomes by capitalizing on the opportunity for early intervention.49
PROGNOSIS
Acute GVHD response rates to high-dose steroid therapy are in the range of 50%, and achievement of response to steroids is the most important predictor of outcomes. Durable responses of a month or longer can be expected in about 30% and partial responses (PRs) in 20%. Lower intestinal acute GVHD has been linked to worse response rates compared to patients without lower intestinal manifestations.
CLINICAL MANIFESTATIONS OF ACUTE GRAFT-VERSUS-HOST DISEASE
The most common sites of involvement are skin, GI tract, and liver. Additional organ sites may be involved and clinicians should maintain a high degree of suspicion for acute GVHD as a cause for any unexplained abnormalities in eyes, buccal mucosa, and lung. A biopsy of the presumptive site should be attempted whenever possible to confirm diagnosis, although histological manifestations can overlap with many inflammatory conditions confounding histological results. A pathologist familiar with acute GVHD is preferable, and appropriate immunohistochemical stains should be employed to rule out viral infection, particularly of the GI tract.
The onset and natural history of acute GVHD is variable depending on organ site, nature, and severity of disease. A hyperacute form, occurring in the first 14 days after transplant,50 is characterized by early onset of typical signs and symptoms, with a preponderance of patients, 90%, exhibiting skin involvement. In addition a higher percentage of patients are likely to experience stage III-IV disease (88% vs. 66%).51, 52
Most cases of acute GVHD manifest within the first 100 days following transplant. With the use of modern prophylaxis regimens including cyclosporine or tacrolimus, the onset is typically 20 to 25 days after cell infusion. With T-cell depletion, average onset is typically at 30 days, although it can be delayed several months as T cells reconstitute.53 NIH consensus criteria for chronic GVHD includes classification of acute GVHD occurring after day 100 (late-onset acute GVHD) and for patients manifesting signs and symptoms of both acute GVHD and chronic GVHD (overlap subtype of chronic GVHD).54 The severity of acute GVHD can easily be quantified using Keystone consensus grading criteria (Table 105.2).
Skin
Skin is the most commonly involved site of acute GVHD, being present 80% of the time at onset. A maculopapular rash is characteristic and may be described as a painful or pruritic sunburn. Characteristically involved sites include the back of the neck, palms, soles, dorsal surfaces of the extremities, and ears, although the rash can spread quickly to include the entire body. Although many cases are mild, severe manifestations can include bullous lesions and a clinical picture consistent with toxic epidermal necrolysis. Careful full-body examination allows for staging of acute GVHD by the Keystone consensus criteria. This scale is based upon percentage of body surface area (BSA) involvement. For example, when acute GVHD is confined to the skin and involves <50% of BSA, it is classified as grade I acute GVHD. Such a degree of involvement may require no more than topical steroids and frequent monitoring of symptoms (see “Primary and Secondary Therapy of Acute Graft-versus-host Disease” below). A biopsy of the skin can help to solidify the diagnosis, however, treatment is usually based on the clinical diagnosis.55
TABLE 105.2 KEYSTONE CONSENSUS CRITERIA STAGING AND GRADING OF ACUTE GRAFT-VERSUS-HOST DISEASE
Skin
Liver
Gut
Stage
1
Rash on <25% of skin
Bilirubin 2-3 mg/dl
Diarrhea >500 ml/d or persistent nausea
2
Rash on 25-50% of skin
Bilirubin 3-6 mg/dl
Diarrhea >1000 ml/d
3
Rash on >50% of skin
Bilirubin 6-15 mg/dl
Diarrhea >1500 ml/d
4
Generalized erythroderma with bullous formation
Bilirubin >15 mg/dl
Severe abdominal pain with or without ileus
Grade
I
Stage 1-2
None
None
II
Stage 3 or
Stage 1 or
Stage 1
III
Stage 2-3 or
Stage 2-4
IV
Stage 4 or
Stage 4
Functional Grading
Skin
Liver
Gut
I
Rash on <50% of skin
II
Rash on <50% of skin or
Bilirubin 2-3 mg/dl or
Diarrhea >500 ml/d or persistent nausea
III-IV
Generalized erythroderma with bullous formation or
Bilirubin >3 mg/dl
Diarrhea >1000 ml/d
Criteria for grading given as minimum degree of organ involvement required to confer that grade.
Grade IV may also involve lesser organ involvement but with extreme decrease in performance status. From Przepiorka D, Weisdorf D, Martin P, et al. 1994 consensus conference on acute GVHD grading. Bone Marrow Transplant 1995;15:825-828.
Hepatic
Liver acute GVHD is typically characterized by involvement of the bile duct epithelium, resulting in cholestasis. Bilirubin and alkaline phosphatase elevation, accompanied by cholestatic jaundice are the typical manifestations. Direct hepatocyte damage is rare, absent a more chronic fibrosis, although transaminitis often occurs. Hyperbilirubinemia must be distinguished from other common post-transplant complications such as toxicity from preparative chemotherapeutics, sinusoidal obstructive syndrome (also called hepatic veno-occlusive disease), and occasionally fulminant viral hepatitis. Sinusoidal obstructive syndrome is characterized by hyperbilirubinemia, portal hypertension, and weight gain due to third spacing of fluids. Portal hypertension is part of the clinical picture of sinusoidal obstructive syndrome, and not a manifestation of acute GVHD in patients without underlying liver disease. Doppler assessment of portal hypertension, measurement of the hepatic vein occlusive pressure, and if necessary histological examination, are critical in resolving the differential diagnosis. A transjugular liver biopsy is preferable to transcutaneous biopsy, as portal pressures can be measured. Acute GVHD produces cholestatic hepatitis, with histology showing frequent acidophilic bodies evolving to bile duct exocytosis and disruption. As the disease progresses beyond day 100 post-transplant, portal fibrosis is seen with increasing bile duct dropout.56 This is in contrast to sinusoidal obstructive syndrome, which is characterized by occluded hepatic venules, sinusoidal fibrosis, and hepatocyte necrosis.57
Gastrointestinal Tract
Anorexia, nausea, and vomiting are the most common symptoms of GI acute GVHD, but diarrhea, abdominal pain, and hemorrhage are symptoms of serious lower-tract disease. Almost any site along the tract can be involved. Unexplained GI symptoms such as mouth ulcers or ileus can be caused by acute GVHD. Endoscopy with biopsies of the upper and/or lower tract should be obtained for persistent symptoms, inasmuch as histology provides critical information. The differential diagnosis of uppertract GVHD includes herpes simplex, cytomegalovirus, or candida esophagitis, stomach ulcer, and peptic gastritis or duodenitis. The differential diagnosis of lower tract disease includes enteritis from C. difficile, cytomegalovirus, Norfolk viruses, cryptosporidium, giardia, or enteric pathogens such as Salmonella, and side effects of irradiation or medications including cytotoxic chemotherapy, tacrolimus, and mycophenolate mofetil among others. Radiologic findings by computed tomography (CT) scan can include thickening of the esophageal, small, or large bowel wall; adjacent vasa recta engorgement; mesenteric fat stranding; or mucosal enhancement.58 Radiologic modalities should not be relied upon for confirmation of diagnosis. Upper or lower endoscopy affords both a visual examination of the mucosa which may exhibit edema, erythema, ulceration, and mucosal sloughing, as well as the opportunity to obtain tissue for histology. Classic microscopic findings include epithelial crypt apoptotic bodies and lymphocytic infiltration. Involved mucosa can be noncontinuous and a lack of findings or a low degree of severity at one level does not rule out other areas or degrees of involvement.59
Lung
Lung complications following allogeneic transplant can be of cardiac, infectious, vascular, or immune nature. Infections and pulmonary embolism should always be entertained in the differential diagnosis of a transplant patient with shortness of breath, hypoxia, or new infiltrate. Volume overload and cardiogenic pulmonary edema is another mechanism of respiratory distress. Immune-mediated processes should be entertained when infectious work-up is negative, or CT scanning is not consistent with infectious processes. Diffuse alveolar hemorrhage has long been recognized as a post-transplant complication and occurs in approximately 10% of patients, with myeloablative regimens associated with a higher incidence. Patients manifest with dyspnea (92%) accompanied with mild to severe bleeding into the alveoli, although hemoptysis is seen in a minority (15%) of cases. Fever can be a presenting symptom (67%) and diagnosis is typically made following bronchoscopy. Patients often require ventilatory support and mortality rates can reach 70%.60, 61 Lung wedge resection or video-assisted thoracoscopic biopsy should be entertained for patients where an organism is not identified and diagnostic uncertainty remains. Bronchiolitis obliterans with organizing pneumonia (BOOP) is seen in up to 2% of patients during the post-transplant period. Although not considered a manifestation of GVHD,62 biopsy-proven BOOP occurs at a median of 108 days after transplant and can display a wide range of severity and reversibility. It is typically a restrictive lung disease with radiographic findings showing peripheral patchy consolidation, ground glass infiltrates, and nodular opacities. BOOP after transplant is associated with GVHD and steroid therapy affords a chance for improvement.63, 64
PREVENTION
A half century ago, Storb and collaborators determined that the immunosuppressive agent methotrexate could mitigate or prevent the onset of acute GVHD, when applied shortly after transplantation. Although oral and liver toxicity can be severe and preclude up to 40% of patients from receiving a full course of therapy, methotrexate remains widely in use, now typically in combination with a calcineurin inhibitor.27 Calcineurin inhibitors act to inhibit IL-2 mediated T-cell expansion. Cyclosporine, the first calcineurin inhibitor, was found to be synergistic with methotrexate, leading to a reduction in severe (grade III/IV) acute GVHD when utilized in HLA-matched sibling allografts and to a lesser degree with unrelated donor allografts.65, 66 Subsequent development of the calcineurin inhibitor, tacrolimus, included two phase III head-to-head comparisons of methotrexate/cyclosporine and methotrexate/tacrolimus.67, 68 The combination of methotrexate/tacrolimus reduced the incidence of grade II-IV acute GVHD for both matched sibling donors (31.9% vs. 44.4%; P = 0.01) and unrelated donor transplants (56% vs. 76%, P = 0.0002), but it did not prevent chronic GVHD or improve survival. This lack of survival advantage propagated the use of both combination regimens at the discretion of individual transplant centers.
Attempts at further decreasing the incidence of acute GVHD have included the addition of agents to a calcineurin inhibitor with or without methotrexate. Mycophenolate mofetil is a prodrug of mycophenolic acid, an inhibitor of de novo synthesis of purines in lymphocytes required for lymphocyte proliferation. Mycophenolate mofetil has been examined in combination with cyclosporine or tacrolimus. Single-center randomized studies of mycophenolate mofetil suggest greater safety but not greater efficacy over methotrexate. One study was stopped early as cyclosporine/mycophenolate mofetil showed a clear advantage over cyclosporine/methotrexate in regard to decreased mucositis (21% vs. 65%, P = 0.008) and faster neutrophil engraftment (11 days vs. 18 days, P < 0.001) but without a difference in the incidence of acute GVHD.69 A second single-center study revealed that mycophenolate mofetil decreased severe mucositis, use of parenteral nutrition, and promoted early hospital discharge, however, mycophenolate mofetil was associated with higher rates of severe (grade III-IV) acute GVHD (19% vs. 4%, p = 0.03), which was predominantly seen with unrelated donors.70
Sirolimus is an mTOR inhibitor that has complex immunomodulatory properties affecting T cells and APC. The drug requires therapeutic monitoring and is affected by inhibitors or inducers of CYP3A4. Sirolimus is associated with the risk of endothelial damage such as in sinusoidal obstructive syndrome.71, 72, 73 Initially shown to be safe in combination with tacrolimus and methotrexate,74 sirolimus has now been tested in several phase II studies in combination with tacrolimus and the two agents likely synergize to reduce acute GVHD rates.75, 76 In particular, sirolimus supports regulatory T-cell reconstitution post-transplant, which are protective against acute GVHD after transplantation.77
Bortezomib is a proteasome inhibitor that has activity against multiple myeloma via nuclear factor kappa B inhibition and other mechanisms. A phase I/II trial showed that bortezomib could be added to tacrolimus and methotrexate as acute GVHD prophylaxis. This regimen is promising as the 180 cumulative incidence of grade II-IV acute GVHD was 13%.78
The chemokine receptor CCR5 plays a role in alloreactivity via effects on lymphocyte migration, and has shown promise in an early phase study. Blockade using the CCR5 antagonist, maraviroc, in combination with tacrolimus and methotrexate as acute GVHD prophylaxis led to inhibition of lymphocyte trafficking, without attenuating T-cell function or impairing stem cell function in vitro.17
Special circumstances surrounding the selection of acute GVHD prophylaxis include HLA-incompatible related donors, unrelated donors, or cord blood. For cord blood transplants, cyclosporine has been the drug primarily utilized as a prophylaxis backbone.79 For HLA mismatched donor transplants, a calcineurin inhibitor/methotrexate-based regimen is generally inadequate for GVHD prophylaxis. Triple combination regimens, ATG, and other methods of T-cell depletion have been shown effective in prevention of acute GVHD for mismatched donor transplants.71, 80
As T cells are the primary effectors of acute GVHD, methods of depleting T cells from the graft or in vivo, following transplant have succeeded in reducing rates and the severity of acute GVHD. An ex vivo method of soybean agglutination and E-rosetting was performed81 to achieve T-cell depletion from allografts, resulting in high rejection rates, presumably due to rejection by residual recipient T cells.82, 83 When combined with more immunosuppressive conditioning regimens this complication is ameliorated, yet even so T-cell depletion has not proven to increase overall survival following transplant, considering increased likelihood for relapse and higher infection rates.84 Similarly, the anti-CD52 monoclonal antibody, alemtuzumab, has been utilized ex vivo to deplete T cells, resulting in low incidence of grade II/IV GVHD (12%), but a concerning rate of graft failure (15%).85 CD34+ cell isolation has been studied and shown to result in extremely low rates of severe acute GVHD in high-risk patients.86
In vivo alemtuzumab has been utilized in combination with several different regimens, both with and without a calcineurin inhibitor, with acceptable rejection rates and low incidence of GVHD.87 The use of alemtuzumab is associated with increased infectious complications.88
Antithymocyte globulin (ATG) refers to polyclonal antibodies against thymus tissue. When used for as prophylaxis or treatment for patients with acute GVHD it leads to depletion of T lymphocytes. It may cause an acute cytokine release syndrome that may include fever, hypotension, and may progress to shock, and delayed “serum sickness.” Infectious complications include Epstein-Barr virus re-activation and associated post-transplant lymphoproliferative disorder, and human herpes virus-6 reactivation and associated encephalitis.89, 90
Another agent used to reduce T-cell numbers following transplant is cyclophosphamide, which is a potent immunosuppressive agent, yet spares stem cells. This strategy can result in low rates of severe acute GVHD, even when used as a single prophylactic agent.91
PRIMARY AND SECONDARY THERAPY OF ACUTE GRAFT-VERSUS-HOST DISEASE
Primary therapy for acute GVHD consists of high-dose steroids (the equivalent of 1 to 2 mg/kg/day of prednisone, with a taper initiated as soon as patients have a major decrease in GVHD signs or symptoms). This standard is based upon historical empirical evidence and randomized controlled data which suggest no advantage to prednisone-equivalent steroid doses of >2.5 mg/kg/day and no disadvantage for 1 mg/kg/day for grade II acute GVHD.92
Standardized consensus response criteria for acute GVHD clinical trials are defined as a complete response, with resolution of all signs and symptoms of disease and normalization of laboratory values, and a very good partial response (VGPR), which allows for residual nonbullous erythematous rash on <25% of BSA, occasional nausea or vomiting, and a reduction of bilirubin to <25% of baseline at enrollment.93 The definition of VGPR (Table 105.3) allows for confounding variables that clinicians must recognize while monitoring for resolution of acute GVHD such as residual GI and hepatic toxicities from conditioning or prophylactic medicines and the recognition of erythema or darkening skin as resolving skin lesions. Prolonged high-dose steroid use leads to a significant degree of morbidity and mortality, and the tapering of these drugs must take precedence once symptomatology and abnormal laboratory data recede.
TABLE 105.3 CONSENSUS TERMINOLOGY DESCRIBING VERY GOOD PARTIAL RESPONSE FOR ACUTE GRAFT-VERSUS-HOST DISEASE
Skin
No rash, or residual erythematous rash involving <25% of the body surface, without bullae (residual faint erythema and hyperpigementation do not count)
Liver
Total serum bilirubin concentration <2 mg/dl or <25% of baseline at enrollment
Gut
Tolerating food or enteral feeding
Predominantly formed stools
No overt gastrointestinal bleeding or abdominal cramping
No more than occasional nausea and vomiting
From Endpoints for Clinical Trials Testing Treatment of Acute Graft-Versus-Host Disease: A Consensus Document. from Martin PJ, Bachier CR, Klingemann HG, et al. Endpoints for clinical trials testing treatment of acute graft-versus-host disease: a joint statement. Biol Blood Marrow Transplant 2009;15:777-784.
Only gold members can continue reading. Log In or Register to continue