Gonadal and extragonadal germ cell tumors (GCTs) are infrequent in childhood, occurring at a rate of 2.4 cases per million children and representing approximately 2% to 3% of cancers diagnosed in children and adolescents younger than 15 years.1 However, GCTs are one of the most prevalent tumors in the adolescent and young adult age group, accounting for 14% of all cancers among those 15 to 19 years. The age distribution is bimodal, and a small peak occurs from infancy to age 4 (type I GCTs), with a second peak occurring after the onset of puberty (type II GCTs),2 with distinct clinical and molecular features in each age group. Building on effective treatments developed for adults with testicular GCTs, clinical trials in pediatric patients with GCTs have led to significant improvements in survival over the last several decades. Future advances in our understanding of the molecular aberrations in these rare pediatric tumors may aid in the development of risk-adapted strategies and further improvements in survival.
NOMENCLATURE
GCTs derive from pluripotent precursor cells known as primordial germ cells (PGCs), and hence can take on a wide array of different histologic appearances. GCTs in which the precursors remain undifferentiated, resembling primitive germ cells, are known as seminomas in males, dysgerminomas in females, and germinomas when occurring in the CNS. GCTs exhibiting differentiation to somatic tissues of the endodermal, mesodermal, and/or ectodermal lineages are known as teratomas. Finally, GCT precursor cells can differentiate to resemble extraembryonic structures such as yolk sac (yolk sac tumor [YST] or endodermal sinus tumor) or placenta (choriocarcinoma [CC]).
EMBRYOGENESIS AND HISTOGENESIS OF GONADAL TUMORS
Analysis of imprinted genetic loci provides some of the strongest evidence that both gonadal and extragonadal GCTs originate from PGCs. Approximately 100 to 200 genes in the human genome are epigenetically imprinted such that only one allele, either maternal or paternal, is normally expressed.3,4 This imprinting is erased in migrating PGCs during embryogenesis, in order to allow sex-specific imprinting during gametogenesis. GCTs typically exhibit partial or complete erasure of imprinting, implying the origin of these tumors from embryonic germ cells.5,6 The histopathologic heterogeneity of GCTs reflects the pluripotent nature of PGCs.7 Further variations with respect to age, sites of presentation, and the malignant potential of GCTs stem from differences in the stage of germ cell development at tumorigenesis, differences in the tumor environment secondary to the gender of the patient and location of the clone, as well as specific genetic aberrations in the tumor. Therefore, understanding the development of embryonic germ cells is critical to an appreciation of these issues.
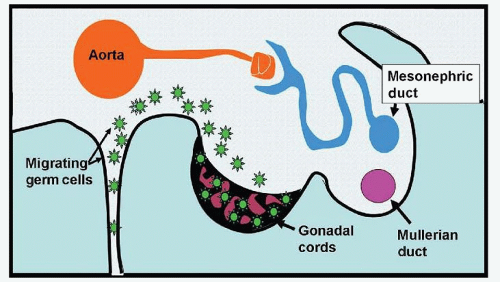
The PGCs first become evident in the extraembryonic yolk sac by the fourth week of gestation. By the fifth week (Fig. 35.1), the germ cells migrate through the mesentery of the hindgut to the gonadal ridge. This migration appears to be mediated by the KIT receptor and its ligand, KITLG, or stem cell factor. PGCs express KIT, and KITLG is expressed with an increasing gradient from yolk sac to the gonadal ridge, guiding germ cells to the gonadal ridge. In animal models, PGC, not expressing KIT, are unable to migrate to the gonad or proliferate during migration.8KITLG was identified as a strong GCT susceptibility locus in several genome-wide association studies,9,10 underscoring the importance of this signaling pathway during germ cell development. Germ cell migration is additionally directed by the chemokine/receptor pair SDF-1/CXCL12 and its receptor CXCR4.11 Germ cells express CXCR4, and migration is directed by the expression and secretion of SDF-1 in the mesenchyme of the gonadal ridge. In the absence of these factors, germ cells do not populate mouse gonads.12
Figure 35.1 Migration of PGCs through mesentery to gonadal ridge. Testicular development is manifested by abrupt development of sex cords. Ovaries do not demonstrate defined sex cords.
The fate of the germ cells after their arrival at the gonadal ridge depends on the sex of the individual. During a narrow window of opportunity in the sixth to seventh week, the Y chromosome-specific gene SRY initiates male sex determination. Ovarian differentiation commences either in the absence of a Y chromosome or if the window of opportunity is missed.13 Testicular differentiation manifests by the abrupt development of cellular cords (sex cords) in the seventh week. PGCs populate these sex cords and then undergo mitotic arrest and remain in that state until puberty. The ovary does not demonstrate the defined sex cords seen in the testis. Instead, immature oocytes arrest in prophase of meiosis I and become surrounded by somatic follicular cells, forming primary follicles. Further development of the oocyte and follicle occurs after puberty with each menstrual cycle.
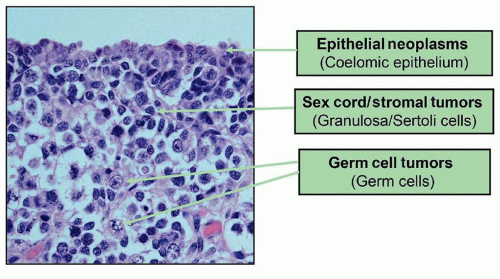
The gonads thus contain three cell types having neoplastic potential (Fig. 35.2). Germ cells give rise to GCTs. The cells of the sex cords may develop into stromal tumors, such as testicular Sertoli or Leydig cell tumors, ovarian granulosa cell tumors, or mixtures of these components. These tumors may sometimes display morphologic features that are discrepant from the sex of the patient, thus illustrating the bisexual differential potential of the gonadal stroma. Finally, coelomic epithelium covering the ovary may evolve into epithelial neoplasms, found most often in adults.
Figure 35.2 Fetal ovary at 18 weeks’ gestation. Coelomic epithelium is responsible for epithelial malignancies. This layer is lost in testicular development, thus low frequency of epithelial tumors in testes. Gonadal stromal tumors arise in a specialized stromal layer. GCTs arise within the PGCs that migrate from yolk sac to gonad early in development.
GENETICS AND MOLECULAR BIOLOGY
Genetic contributions to the pathogenesis of pediatric GCTs include germline genetic changes leading to increased susceptibility and tumor-specific (somatic) genetic changes. Familial tumors appear to account for 1.5% to 2% of all adult GCTs,14 and it is likely that a similar proportion of adolescent GCTs are heritable. Whether some type I infantile GCTs also arise from an inherited predisposition is not known. The association between sex chromosome abnormalities and the development of GCTs is well established. In particular, individuals with 46, XY and 45, X/46, XY gonadal dysgenesis have a 10% to 50% risk of developing a gonadal GCT. Patients with Klinefelter syndrome (47, XXY) have an increased risk of developing extragonadal GCTs, especially mediastinal GCTs.15 In addition, the average age at GCT diagnosis in Klinefelter patients is 10 years younger than in those with a normal constitutional karyotype. In one study, 50% of adolescents with mediastinal GCTs had cytogenetic changes consistent with Klinefelter syndrome.16 Patients with mixed gonadal dysgenesis with ambiguous genitalia, or 45, X Turner syndrome with retained Y chromosome material, are at risk of developing gonadoblastoma, a mixed germ cell-sex cord-stromal tumor.17
Genetic Characteristics of Testicular Tumors in Adolescents and Adults
Overrepresentation of chromosome 12p is a universal feature of adolescent/adult GCTs.18 An isochromosome 12p (i[12p]) is pathognomonic of GCT and can be detected cytogenetically in 80% of cases. In other cases, an increased copy number of 12p sequences can be detected. The amplicon 12p11-12p12.1 is present in many adolescent/adult GCTs and includes potentially disease-relevant genes such as KRAS, CCND2, NANOG, and DPPA3. However, the exact contribution of 12p amplification to GCT development is not known.19,20
In addition, loss of all or parts of chromosomes 4, 5, 11, 13, 18, and Y and gain of 7, 8, 12, and X is a common finding in adolescent and adults GCTs.21,22 Recently, a study of genome-wide copy number variation (CNV) in seminomas, using high-resolution single-nucleotide polymorphism arrays, confirmed many previously reported findings and identified novel CNVs, including both amplifications and homozygous deletions,23 including potential candidate seminoma susceptibility genes such as FAT3, EDNRB1, and RASSF8.
Additional molecular studies have shed further light on adolescent/adult GCT pathogenesis. Activating mutations of KIT and RAS have been reported in testicular GCTs.24 A number of studies have profiled mRNA expression in testicular GCTs in adult men, revealing histologic subtype-specific gene expression patterns.25,26 Korkola and coworkers used microarrays to identify and validate a prognostic mRNA gene expression signature that defined 5-year overall survival in adult nonseminomatous TGCTs.27
Genetic Characteristics of Ovarian Tumors in Adolescents and Adults
The genetic biology of ovarian GCTs is more complex than that of testicular GCTs and is considered separately for mature teratomas, immature teratomas, and malignant GCTs.
Mature and Immature Teratomas
Cytogenetic assessment of more than 325 ovarian mature teratomas demonstrated that 95% were karyotypically balanced, with only 5% showing heterogeneous gains of single whole chromosomes. Studies of molecular loci have shown that the majority of mature ovarian teratomas have entered but not completed meiosis. These diploid tumors are genetically unique in that the majority of their genome is isodisomic, i.e., both chromosomes in a pair are from one parent only. Accordingly, these teratomas may show a methylation profile of imprinted genes (e.g., hypermethylation of the small nuclear ribonucleoprotein N gene), consistent with a postmeiotic origin.6
Ovarian immature teratomas are heterogeneous. Some show evidence of a meiotic stem cell origin, and others show mitotic origins, suggesting the failure of early meiotic arrest. This implies that immature and mature teratomas may represent different biologic entities, rather than simply a spectrum of maturation. The frequency of chromosomal abnormalities in immature teratoma is higher than in mature teratoma. Most patients with cytogenetically abnormal immature teratomas reported to date have experienced multiple recurrences. In contrast, patients with karyotypically normal immature teratomas have remained disease free.
Malignant Ovarian GCTs
Malignant ovarian GCTs show similar ploidy and genetic features when compared with their testicular counterparts. They are aneuploid: approximately 75% contain i (12p); 42% and 32% have gains of chromosomes 21 and 1q, respectively; 25% and 42% have loss of chromosomes 13 and 8, respectively.
In summary, although malignant ovarian GCTs appear to be equivalent to their adolescent testicular counterparts, immature and mature ovarian teratomas remain unique subcategories of GCTs likely to have a different mechanism of origin. Immature teratomas may develop genetic changes that are accompanied by histologic malignant transformations.
Genetic Characteristics of Extragonadal GCTs of Older Children
During embryogenesis, PGCs migrate through midline structures, suggesting that extragonadal GCTs arising in the midline may have their origin in aberrantly migrated germ cells.28 However, direct experimental evidence for this hypothesis is lacking. Ectopic germ cells only rarely have been reported to exist in human embryos, most having disappeared by 18 weeks’ gestation. Similar to testicular GCTs, extragonadal GCTs in older children, which most commonly occur in the mediastinum and brain, show the absence of methylation of most imprinted genes, strongly supporting a germ cell origin.6 Ploidy analyses of mediastinal GCTs suggest that most are diploid or tetraploid. Those that are malignant contain the i (12p) and the other genetic changes seen in adolescent testicular GCTs.16 Recently, integrated genomic analysis of intracranial GCTs identified recurrent genetic lesions resulting in activation of KIT/RAS and PI3 kinase/mTOR signaling in these tumors, along with mutations in the transcriptional repressor BCORL1 and enrichment of novel, rare germline variants of the histone demethylase, JMJD1C.29 The extragonadal GCTs in adolescents and adults are associated with hematopoietic malignancies of various cell lineages that present soon after the initial presentation of the GCTs. The malignant hematopoietic clone commonly demonstrates i (12p), unlike hematopoietic malignancies that arise secondary to therapy.
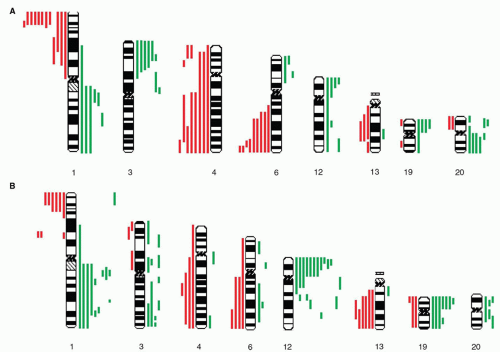
Figure 35.3 Array comparative genomic hybridization of GCTs in children and adolescents. Selected chromosomes are shown; vertical green bars indicate gains in individual tumors and red bars indicate losses. A: summary karyograms for children <5 years of age. Note the presence of frequent losses of chromosomes 1p, 4, and 6q and the occurrence of four tumors with amplification of chromosome 12p. B: summary karyograms for children 5 and <16 years of age. The predominant copy number imbalance is gain of chromosome 12p; also notable are gains at 1q and 19, and losses at chromosomes 1p, 6q, and 13. (Adapted by permission from Macmillan Publishers Ltd: Palmer RD, Foster NA, Vowler SL, et al. Malignant germ cell tumours of childhood: new associations of genomic imbalance. Br J Cancer 2007;96(4):667-676.)
Genetic Characteristics of Extragonadal and Testicular GCTs of Young Children
In children younger than 4 years, GCTs arising in gonadal and extragonadal sites are histologically, clinically, and genetically similar. Most teratomas in this age group are diploid, have normal karyotypes, and, if completely resected, behave in a benign fashion regardless of degree of immaturity and site of origin. Malignant GCTs in these young children are almost exclusively YSTs that demonstrate only partial erasure of imprinting, suggesting that the tumors arise from an earlier stage of germ cell development than do adolescent/adult GCTs.6 Recurrent cytogenetic abnormalities include gains at chromosomes 1q and 20q, and losses at chromosome 1p and chromosome 6q, findings that were confirmed in a larger study from the UK Children’s Cancer and Leukaemia Group (CCLG; Fig. 35.3).30
Palmer and coworkers conducted gene expression profiling of pediatric GCTs, identifying a self-renewal/pluripotency gene expression signature in seminomas, and a gene signature associated with differentiation and proliferation in YSTs.31 This study further demonstrated that adult and pediatric GCTs exhibit distinct gene expression patterns, even within the same histologic subclass. Taken together with the differences in genomic imprinting and the overall spectrum of chromosomal changes, these data support a model whereby infantile and adolescent/adult GCTs arise from distinct populations of embryonic germ cells. However, it is worth noting that the CCLG study documented gain of chromosome 12p in more than one-third of GCTs from children less than 5 years of age.30 Thus, infantile and adolescent/adult malignant GCTs may in fact share certain pathogenic mechanisms.
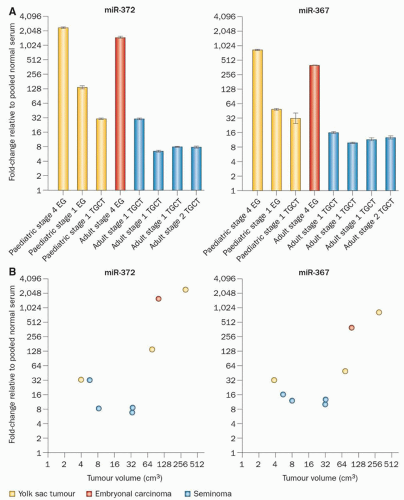
Figure 35.4 Micro-RNAs are potential serum biomarkers of GCT. A: Expression of miRNAs miR-372 (left) and miR-367 (right) in the serum of GCT patients at diagnosis, compared to levels in control sera. miR-372 and miR-367 are uniformly elevated in GCT irrespective of age, tumor site, or tumor histology. B: Correlation between serum level of miR-372 and miR-367 at diagnosis with volume of the primary tumor. (Adapted by permission from Macmillan Publishers Ltd: Murray MJ, Coleman N. Testicular cancer: a new generation of biomarkers for malignant germ cell tumours. Nat Rev Urol 2012;9(6):298-300.)
The Role of Noncoding RNAs in GCTs
MicroRNAs (miRNAs) are approximately 22-nt noncoding RNAs that regulate the stability and translation of target mRNAs.32,33 miRNA-372 and miRNA-373 were first demonstrated as oncogenes in testicular GCTs,34 and further studies established that both gonadal and extragonadal GCTs uniformly overexpress miRNAs from the miRNA-371˜373 and miRNA302 clusters.35 While the mRNA targets of these miRNAs may prove to be relevant to GCT pathogenesis or the target for novel therapeutic approaches,36 the more immediate translational significance of this finding derives from the fact that miRNAs are shed from tumor cells into the circulation,37 and thus may prove to be biomarkers for GCTs that do not secrete α-fetoprotein (AFP) or human chorionic gonadotropin (HCG). Indeed, serum levels of miRNA-371˜373 and miRNA302 are increased in GCT patients at diagnosis, regardless of age, tumor histology, or location (Fig. 35.4).38,39
PATHOLOGY
GCTs are composed of numerous histologic subtypes that are independent of presenting clinical characteristics, tumor biology, or clinical behavior, and vary with site of origin, stage, and age of the patient. For example, mature teratomas in infants are almost invariably diploid and benign, whereas those in the adult testis, with the same histologic features, are aneuploid and potentially malignant. The histologic classification of these tumors is shown in Table 35.1.
Teratoma
Teratomas, the most common histologic subtype of childhood GCT, are classified according to their histologic composition as mature, containing well-differentiated tissues; immature, containing varying degrees of immature fetal tissue, most often neuroectodermal; or malignant, containing at least one of the malignant germ cell elements. Mature teratomas of the gonads are encapsulated, multicystic, or solid. Extragonadal teratomas differ from those arising from the gonads only in the absence of a clearly defined external capsule.
TABLE 35.1 Histologic Classification of Pediatric Gonadal and Extragonadal Tumors
Ovarian
Germ cell
Teratoma
Mature (solid, cystic)
Immature
1. Mature tissue only
2. Immature tissue, less than 1 low-power field per slide
3. Immature tissue, 1-3 low-power fields per slide
The mature teratoma is composed of representative tissues from all three germ cell layers (ecto-, meso-, and endoderm). Although any tissue type may be present, the most common are skin and skin appendages, adipose tissue, mature brain, intestinal epithelium, and cystic structures lined by squamous, cuboidal, or flattened epithelium. Hematopoietic, pancreatic, or pituitary tissue is frequently found in mediastinal tumors and rarely in teratomas at other sites.
Immature teratomas primarily occur in extragonadal sites in children or in the ovaries of girls near puberty.40 Although their gross appearance is similar to mature teratomas, the presence of various immature tissues, usually neuroepithelial, is a unique feature. A number of grading systems for immature teratoma, all of which are variations of the system originally devised by Thurlbeck and Scully,41 quantify the degree of immaturity in the lesion (Table 35.1). The risk of local recurrence is higher in immature teratomas, particularly for those in the sacrococcygeal region, mostly due to a higher proportion of incomplete resections rather than the degree of immaturity. Immature teratomas in children behave in a malignant fashion, only if foci of malignant germ cell elements (usually YST) and specific clinical characteristics (usually advanced stage) are present. However, if resected completely, the presence of malignant YST foci does not impact on prognosis. Clusters of YSTs can be easily overlooked because they may be very small, associated intimately with the immature neural tissue, and they frequently do not stain positively for AFP. Tumors containing such foci are likely responsible for the reports that immature teratoma may metastasize.
Yolk Sac Tumor
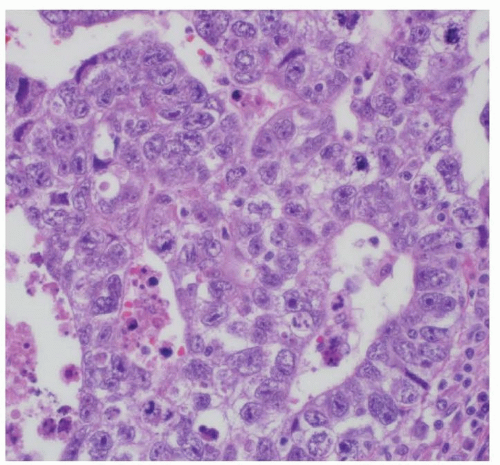
YSTs are the most common pure malignant GCT in young children as well as the most common testicular GCT, benign or malignant, in infants and young boys. YSTs are the only malignant GCT type occurring in the sacrococcygeal region of infants. In adolescents, extragonadal YSTs rarely occur in pure form, but, more frequently, are a component of a mixed malignant GCT. These tumors consist of friable, pale gray, mucoid tissue in which variable amounts of hemorrhage and necrosis are present. The microscopic features are varied and have been characterized fully only in the last two decades.42 Individual cells may be small and pale, with scant cytoplasm, round to oval nuclei, and unapparent nucleoli, or they may be medium sized to large with clear vesicular nuclei and prominent nucleoli, resembling cells of an embryonal carcinoma or germinoma. Mitoses range from few to many.
Four general patterns and a number of variations have been recognized, including pseudopapillary, reticular, parietal, and solid. These patterns are useful in the recognition of YSTs but have no other known clinical relevance (Fig. 35.5).
Germinoma
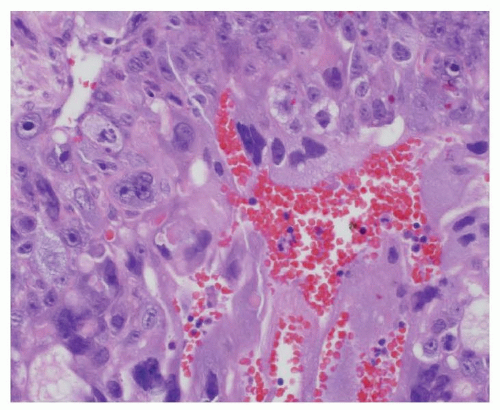
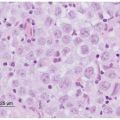
Germinomas, also termed dysgerminomas (ovary) or seminomas (testis), are the most common pure malignant GCTs that occur in the ovary and central nervous system in children. Pure seminomas, which are the most common malignant GCT in men older than 20 years, are unusual in younger males with the exception of those with sex-chromosomal abnormalities or cryptorchidism. Grossly, germinomas are encapsulated, solid, gray-pink tumors with a rubbery consistency and occasional small foci of hemorrhage or necrosis. Microscopically, the tumor cells are arranged in nests separated by bands of fibrous tissue containing variable numbers of lymphocytes (Fig. 35.6). The cells are large, with clear cytoplasm, distinct cell membranes, and large round nuclei having one or two prominent nucleoli. Granulomas with giant cells are frequently present. Syncytiotrophoblasts may also be present, but they are not of prognostic significance, unless they are associated with cytotrophoblasts in foci of CC. Immunohistochemically, the germinoma cells stain for placental alkaline phosphatase (PLAP) and c-kit, whereas the syncytiotrophoblasts stain for HCG beta-subunit (β-HCG). In addition, the stem cell marker OCT-4, which stains positive in germinomas and embryonal carcinomas, has proven useful in the immunohistochemical assessment of germinomatous lesions, in particular in the differential distinction of testicular intratubular neoplasia.43
Figure 35.5 Yolk sac tumor. Schiller-Duval body is characterized by central blood vessel closely invested by a layer of tumor cells and separated from a second layer of tumor cells by a space, likened by some, to Bowman’s space in glomerulus.
Figure 35.6 Germinoma (seminoma). Nests of monomorphic cells with abundant clear cytoplasm and round to oval vesicular nuclei with prominent nucleoli are separated by bands of connective tissue.
Embryonal Carcinoma
Embryonal carcinoma is most commonly a component of a mixed GCT, rather than a primary histopathologic subtype. This component is seen in adolescent testicular GCTs. They are characterized by large cells with large, overlapping nuclei and very large, round nucleoli (Fig. 35.7). The major pattern is epithelial and consists of large nests of cells with varying amounts of central necrosis. Pseudotubular and papillary patterns that may be confused with those of YSTs are frequent, but the cells are AFP-negative, and the tumors typically lack the eosinophilic hyaline globules characteristic of YSTs. They stain positive for OCT-4, and unlike other GCTs, embryonal carcinoma is positive for CD30 by immunohistochemical staining.
Choriocarcinoma
Like embryonal carcinoma, CC rarely occurs outside the context of malignant mixed GCTs in adolescents. The rare case of pure CC detected in infants almost always represents metastasis from maternal or placental gestational trophoblastic primary tumor. These tumors are characteristically very hemorrhagic and friable. Microscopically, two types of cells must be present to confirm the diagnosis: cytotrophoblasts, which classically appear as closely packed nests of relatively uniform, medium-sized cells having clear cytoplasm, distinct cell margins, and vesicular nuclei, and syncytiotrophoblasts, which represent multinucleate syncytial trophoblastic cells (Fig. 35.8). The syncytiotrophoblastic elements stain positively for β-HCG, accounting for the associated high concentrations of serum β-HCG in these patients.
Gonadoblastoma
Gonadoblastoma is a benign tumor found in dysgenetic gonads of phenotypic female subjects who have at least a portion of the Y chromosome. The tumors are usually small (1 to 3 cm in diameter), soft to firm, gray-tan to brown, and slightly lobulated. They are often gritty on cut sections because of the presence of multi-focal calcifications. Microscopic features include the proliferation of both germ cells, positive for PLAP and OCT-4, and gonadal sex cord cells. Germinoma frequently develops with gonadoblastoma. In addition, because gonadal dysgenesis may not be recognized until adolescence, the presence of calcifications within a dysgerminoma of an otherwise-normal female patient suggests the prior presence of gonadoblastoma and, hence, merits evaluation of the patient for gonadal dysgenesis.
Figure 35.7 Embryonal carcinoma. Large, vesicular, overlapping nuclei with large prominent nucleoli and moderate amounts of eosinophilic cytoplasm are seen. Necrotic cells, typical of this histology, are present.
Figure 35.8 CC. Nests of small cells with clear cytoplasm, round vesicular nuclei, and prominent nucleoli (cytotrophoblasts) are intimately associated with large, giant syncytiotrophoblasts.
Associated Pathologic Findings
Intratubular Germ Cell Neoplasia
Seminiferous tubules adjacent to testicular malignant GCTs in adolescents and adults may show increased numbers of enlarged, atypical germ cells with abundant clear cytoplasm and prominent nucleoli. These cells show positivity for PLAP, c-kit, and OCT-4, similar to the cells of seminomas. Such foci have been termed intratubular germ cell neoplasias, and are thought to represent neoplasia in situ. The germ cells in the seminiferous tubules adjacent to infantile testicular malignant GCTs may also be somewhat increased in number and slightly enlarged, with abundant, clear cytoplasm. These cells are negative for the markers of intratubular neoplasia, OCT-4, PLAP, and c-kit, and therefore do not qualify as neoplastic precursor lesions.
Gliomatosis Peritonei
Many ovarian teratomas are associated with nodules of mature glial tissue implanted throughout the peritoneum or in lymph nodes. Mature glial nodules have also been described in cervical lymph nodes in association with teratomas of the head and neck. If these tissues are mature and composed only of glial tissue, this process is termed gliomatosis peritonei, and neither tumor stage nor prognosis is affected. It is not known if immature neural or other mature or immature nonneural tissues are associated with the same benign prognosis. Gliomatosis peritonei may rarely be detected in the absence of malignancy. Accordingly, there is evidence that the glial tissue is reactive rather than neoplastic and does not belong to the same clonal origin as the teratoma.
TUMOR MARKERS
Serum tumor markers, the “onco”-fetoproteins (AFP and β-HCG), aid in the diagnosis of GCTs, as well as in the detection of residual or progressive disease. In adult men with testicular cancer, elevations in these markers were shown to portend a poor prognosis,44 and rate of decline has been demonstrated to be a sensitive early marker of response to therapy.45 Initial results of a risk classification study in pediatric and adolescent GCTs do not show a correlation between AFP and prognosis46 or response to therapy; although, as explained below, the interpretation of AFP is less straightforward in children due to the age-dependent variation in normal levels.
α-Fetoprotein
AFP, an α1-globulin, is the earliest and predominant serum-binding protein in the fetus, reaching its peak concentration at 12 to 14 weeks of gestation and gradually falling to reach an adult normal level of less than 10 ng/dL at approximately age 1 year. As AFP levels begin to decline in fetal development, albumin becomes the principal serum-binding protein. In early embryogenesis, AFP is produced in the yolk sac and later by hepatocytes and the gastrointestinal tract. Elevated serum levels or positive immunohistochemical staining of GCTs for AFP indicates the presence of malignant components, specifically yolk sac or embryonal carcinoma. The serum half-life (t1/2) of AFP is 5 to 7 days. Because of the wide variation in levels at birth, especially with infants of less than 40 weeks’ gestational age, and the wide variability in t1/2 at different ages within the first year of life, difficulties arise in interpreting both the absolute level of AFP in the first year of life and the decay of serum AFP as an indication of residual or recurrent GCT in infants younger than 8 months. Normal ranges have been established to address these problems (Table 35.2).47,48
TABLE 35.2 Normal Ranges of Serum α-Fetoprotein in Infants
WU ET AL.
BLOHM ET AL.
Age
No. of Patients
Mean ± SD (ng/mL)
Age
Mean (95% CI)
Premature
11
134,734 ± 41,444
Premature
158,125 (31,261-799,834)
Newborn
55
48,406 ± 34,718
Newborn
41,687 (9,120-190,546)
Newborn-2 wk
16
33,113 ± 32,503
Day 8-14
9,333 (1,480-58,887)
2 wk-1 mo
12
9,452 ± 12,610
Day 15-28
1,396 (316-6,310)
2 mo
40
323 ± 278
Day 46-60
178 (16-1,995)
3 mo
5
88 ± 87
Day 61-90
80 (6-1,045)
4 mo
31
74 ± 56
Day 91-120
36 (3-417)
5 mo
6
46.5 ± 19.0
Day 120-150
20 (2-216)
6 mo
9
12.5 ± 9.8
Day 151-180
13 (1.25-129)
7 mo
5
9.7 ± 7.1
8 mo
3
8.5 ± 5.5
Day 181-720
8 (0.8-87)
From Wu JT, Sudar K. Serum AFP levels in normal infants. Pediatr Res 1981;15:50, with permission; and Blohm MEG, Vesterling-Horner D, Calaminus G, et al. Alpha 1-fetoprotein (AFP) reference values in infants up to 2 years of age. Pediatr Hematol Oncol 1998;15(2):135-142.
Increasing levels of serum AFP usually indicate either residual tumor after surgery or tumor progression. However, there are other potential explanations for rising or persistent AFP levels. In children, especially very young children, there may be a slow rise in AFP that is observed after completion of therapy; hence, on the last COG protocol, AGCT0132, a rise in tumor markers to more than 5 times the normal value was required to declare relapsed disease. An abrupt escalation in serum AFP has also been described when chemotherapy is first initiated, likely reflecting chemotherapy-induced tumor lysis.49 Spurious persistence of elevated serum AFP may reflect an alteration in hepatic function from conditions such as viral hepatitis (hepatitis B, hepatitis C, and human immunodeficiency virus-associated hepatitis), cholestasis secondary to anesthesia, or exposure to phenytoin or methotrexate.49,50 Other conditions associated with elevated serum AFP include hepatoblastoma, pancreatic and gastrointestinal malignancies, lung cancers, and benign liver conditions, including hepatic dysfunction and cirrhosis.51
β-Subunit of HCG
HCG, a glycoprotein composed of α- and β-peptide subunits, is normally synthesized during pregnancy by syncytiotrophoblasts of the placenta to maintain viability of the corpus luteum. The α-subunit is similar to α-peptides of other hormones, such as luteinizing hormone, follicle-stimulating hormone, and thyroid-stimulating hormone; the β-subunit is antigenically distinct, serving as the basis for the method of serum assays. Minute amounts of β-HCG, less than 5 IU/mL, are detected in serum of healthy adults; serum t1/2 of β-HCG is 24 to 36 hours. Elevation of serum β-HCG in patients with GCTs implies the presence of clones of syncytiotrophoblasts, such as CC, or of syncytiotrophoblastic giant cells, found frequently in germinomas (pure seminomas or dysgerminomas) and occasionally in adult embryonal carcinoma.
Rising or persistently elevated levels of β-HCG usually indicate residual or progressive disease, but, as with AFP other explanations, may exist and warrant investigation. Iatrogenic hypogonadism secondary to bilateral orchiectomy, oophorectomy, or chemotherapy can be associated with rising levels of serum β-HCG because of an increase in luteinizing hormone that results in immunologic cross-reactivity.50 Other conditions in which modest elevations of serum β-HCG have been reported include multiple myeloma and other malignancies of the liver, pancreas, gastrointestinal tract, breast, lung, or bladder.
Other Markers
Because up to 50% of GCTs with identifiable malignant elements do not produce measurable amounts of serum AFP or β-HCG, other markers with potential prognostic value have been investigated. Serum LDH, a glycolytic enzyme that appears to correlate with growth and regression of various solid neoplasms, has not shown specificity for a specific histologic subtype of GCTs. In children with pediatric GCTs, the Brazilian Germ Cell Tumor Group reported that elevation of LDH more than 1.5 times normal was a strong predictor of relapse52; this finding deserves prospective validation in other clinical trials.
Sex cord-stromal tumors may be associated with elevated levels of inhibin, a marker of steroid hormone production. Serum hormone levels, such as estrogen and testosterone, may also be elevated and can serve as both a diagnostic aid and a means of detecting relapse or progression.
CLINICAL PRESENTATION AND STAGING
Ovarian Tumors
Ovarian tumors are rare prior to the onset of puberty. The incidence of ovarian GCTs begins to rise around age 8 and increases throughout young adulthood. Two-thirds of pediatric and adolescent ovarian tumors are of germ cell origin with tumors of epithelial and stromal origin occurring less frequently.53
TABLE 35.3 Children’s Oncology Group Staging of Testicular, Ovarian, and Extragonadal Tumors
Testicular
I
Limited to testis, completely resected by high inguinal orchiectomy; no clinic, radiographic, or histologic evidence of disease beyond the testis; tumor markers normal after appropriate half-life decline; patients with normal or unknown markers at diagnosis must have negative ipsilateral retroperitoneal lymph node sampling to confirm stage I disease
II
Transcrotal orchiectomy; microscopic disease in scrotum or high in spermatic cord (<5 cm from proximal end); retroperitoneal lymph node involvement (<2 cm) and/or increased tumor markers after appropriate half-life decline
III
Tumor-positive retroperitoneal lymph node(s) >2 cm diameter; no visceral or extra-abdominal involvement
IV
Distant metastases that may include liver
Ovarian
I
Limited to ovary, peritoneal washings negative for malignant cells; no clinical, radiologic, or histologic evidence of disease beyond the ovaries (gliomatosis peritonei did not result in upstaging); tumor markers negative after appropriate half-life decline
II
Microscopic residual or positive lymph nodes (<2 cm); peritoneal washings negative for malignant cells (gliomatosis peritonei did not result in upstaging); tumor markers negative or positive
III
Gross residual or biopsy only, tumor-positive lymph node(s) >2 cm diameter; contiguous visceral involvement (omentum, intestine, bladder); peritoneal washings positive for malignant cells
IV
Distant metastases that may include liver
Extragonadal
I
Complete resection at any site, coccygectomy included as management for sacrococcygeal site, negative tumor margins
II
Microscopic residual; lymph nodes negative
III
Gross residual or biopsy only; regional lymph nodes negative or positive
IV
Distant metastases that may include liver
Abdominal pain is the presenting symptom in up to 80% of patients.54 The pain can be chronic, but may mimic an acute abdomen. Most latter cases have associated ovarian torsion and often undergo exploratory surgery for presumed acute appendicitis. Other presenting signs and symptoms include a palpable abdominal mass, fever, constipation, amenorrhea, vaginal bleeding, and rarely frequency and dysuria. Precocious puberty, more often associated with ovarian sex cord-stromal tumors, has also been described in YSTs, CCs, and mixed teratomas with sarcomatous and non-germ cell carcinomatous elements.
Ultrasound, which is most often used for the initial evaluation of patients with abdominal or pelvic masses, will differentiate cystic from solid masses. The presence of a solid ovarian mass raises the suspicion of malignancy. Computed tomography (CT) or MRI of the abdomen and pelvis is helpful in identifying the site of origin, the extent of tumor, the presence of calcifications or fat, and metastatic disease. Staging should also include a chest CT. Metastases to bone, bone marrow, or brain are uncommon, and investigation of these sites is only indicated in patients with clinical indications, stage IV disease, or CC.
A modification of the FIGO staging system was devised by the Pediatric Oncology Group (POG) and Children’s Cancer Group (CCG) for intergroup studies (Table 35.3). This surgicopathologic system refines the FIGO system by accounting for (a) the higher risk of tumor recurrence in patients with positive peritoneal washing who are therefore upstaged from FIGO IC to COG stage III, and (b) the lack of negative prognostic implications of gliomatosis peritonei if only mature glial tissue is present. In all ovarian tumors, cytologic evaluation of ascitic fluid or a peritoneal washing is mandatory. Clinical features to various ovarian tumors are summarized below.
Presentation of Ovarian GCTs as a Function of Histology
The mature cystic teratoma, or dermoid tumor, is the most common type of GCT, and like all ovarian GCTs is most common during the second and third decades of life. Approximately 10% of patients with teratomas have bilateral tumors and, in this instance, every effort should be made to preserve fertility. Dysgerminoma is also often bilateral, in up to 20% of cases. Pure YSTs are the most common histology in malignant pediatric GCTs and are associated with an elevation in AFP in almost all cases. In the postpubertal patient, a tumor of mixed histologies is most prevalent. Patient with embryonal carcinoma and CC can present with symptoms of precocious puberty, amenorrhea, or hirsutism, due to production of β-HCG by the multinucleated giant cells in the tumor. These patients are not infrequently misdiagnosed as pregnant because of the false-positive pregnancy test, which compounds the emotional distress of patient and family.
Presentation of Gonadoblastoma
Gonadoblastomas are tumors composed of germ cells intermixed with stromal cells (usually Sertoli or granulosa cells, with or without Leydig cells). Most gonadoblastomas are small-to-medium sized and behave in a benign fashion unless there is overgrowth of a malignant germ cell element. Although most tumors are unilateral, up to 36% are bilateral. These neoplasms develop during the teenage years, most frequently in patients with XY gonadal dysgenesis, although a small number may occur in patients with 45, XO/46, and XY mosaicism. Patients are usually seen for evaluation of amenorrhea, invariably leading to an ultrasound that is diagnostic. Patients may have a eunuchoid body habitus, elevated gonadotropin levels, and streak gonads.
Presentation of Sex Cord-Stromal Tumors
Granulosa cell tumors have two distinct forms: juvenile and adult.55,56 Juvenile granulosa cell tumors are seen in young children and often present with precocious puberty. The vast majority are localized (i.e., stage I) at diagnosis and associated with a favorable prognosis. Juvenile granulosa cell tumors rarely recur, and when this occurs, it is generally within the first 2 to 3 years after diagnosis. Adult granulosa cell tumors are very rare; less than 5% of cases occur in children. Adult granulosa cell tumors have a propensity for late relapse, often more than 10 years after diagnosis. Granulosa cell tumors of both types may secrete inhibin, which is a valuable diagnostic marker.
Only gold members can continue reading. Log In or Register to continue