Genomic Stability and DNA Repair
5.1 INTRODUCTION
There is overwhelming evidence that mutations can cause cancer. Major evidence for the genetic origin of cancer includes: (a) the observation of Ames (Ames et al, 1981) that many carcinogens are also mutagens, and (b), the finding that genetically determined traits associated with a deficiency in the enzymes necessary to repair lesions in DNA are associated with an increased risk of cancer. Mutations may occur in the germline of an individual and be represented in every cell in the body, or they may occur in a single somatic cell and be identified in a tumor following clonal proliferation. As described in Chapter 6, all species have numerous genes called cellular oncogenes (or protooncogenes), many of which are homologous to the transforming oncogenes carried by specific RNA retroviruses. Some human tumors have mutations in these oncogenes that may have led to their activation. However, there is no evidence for germline mutations in cellular oncogenes, perhaps because such mutations in the germline are lethal even in the heterozygous state. In contrast, there is good evidence for germline mutations affecting tumor-suppressor genes, which can lead to familial clustering of cancer or transmission of predisposition to tumors. In such cases, the loss-of-function of a tumor-suppressor gene is inherited in a mendelian manner.
The maintenance of genetic information is of paramount importance for prevention of genetic instability and accompanying carcinogenesis. In this chapter, intrinsic and extrinsic causes of genomic instability are discussed and the biochemical pathways that act to repair specific DNA lesions and the function of cell-cycle checkpoints following DNA damage are described. The methods used to evaluate DNA damage sensing and repair are detailed and human disorders that result in defective DNA damage sensing and repair are discussed. Finally, the importance of the appropriate maintenance of chromosomal length and telomerase activity is highlighted. Throughout the chapter, examples are given of the importance of each of these factors in the genesis, diagnosis, and treatment of human cancer.
5.2 GENETIC INSTABILITY AS THE BASIS FOR MALIGNANT TRANSFORMATION
5.2.1 Intrinsic Causes of Genetic Instability
Cellular carcinogenesis is known to require sequential mutations in DNA (see Chap. 4, Sec. 4.2.3). Damaged DNA is produced by a number of mechanisms, including (a) spontaneous reactions of DNA with the aqueous environment, (b) influence of metabolic byproducts such as reactive oxygen or nitrogen species, (c) action of environmental mutagens such as radon and chemical exposures, and (d) errors during DNA replication. Together these mechanisms can produce abasic sites, deamination, base alterations, single-strand DNA breaks and double-strand DNA breaks, which can amount to as many as 105 DNA lesions per cell per day (Ciccia and Elledge, 2010; Hoeijmakers, 2009). Unless the cell can protect and maintain the integrity of the genome, these genetic alterations may cause cancer by activating protooncogenes and/or inactivating tumor-suppressor genes. Natural mutation rates appear sufficient to drive the selection required for formation of many tumors (Bodmer et al, 2008). Some inactivating mutations occur in genes responsible for maintaining genomic integrity or DNA repair, which facilitate the development of a mutator cellular phenotype (Fig. 5–1; Loeb, 1991). Regardless of the nature of the genotoxic insult, genomic instability is believed to be the main “enabler” of malignant transformation.
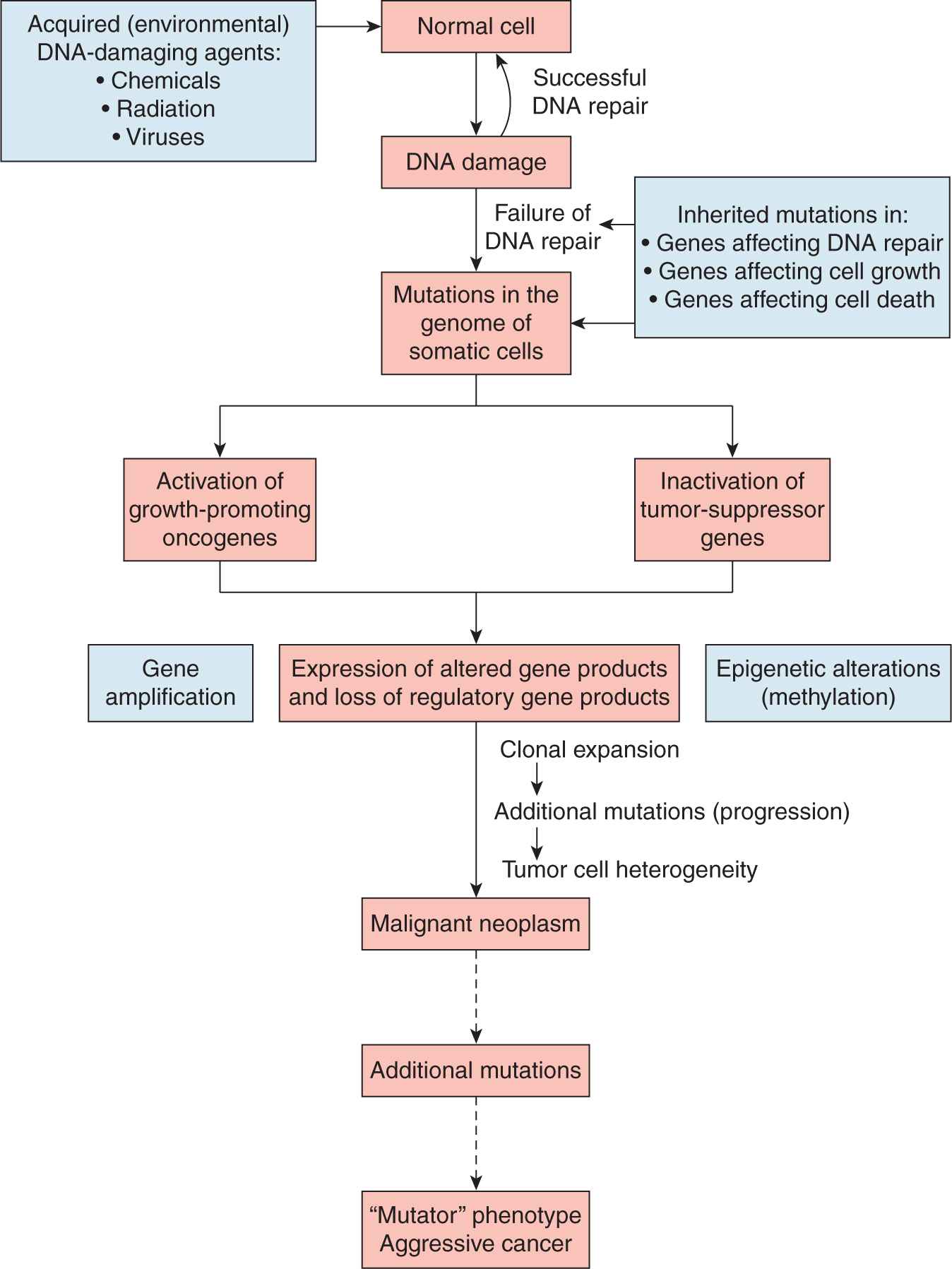
FIGURE 5–1 Genetic and epigenetic mechanisms of genetic instability.
There are mechanisms other than mutation that lead to genetic instability, including gene amplification, and the epigenetic modification of chromatin-associated, transcriptional states through gene methylation and gene acetylation (see also Chap. 7, Sec. 7.4). In cells that display gene amplification, gene expression is aberrantly and constitutively enhanced above the level of physiologically normal levels. Gene-amplified cells can display discrete cytogenetic changes, such as double-minute (DM) chromosomes or homogeneously staining regions (HSRs). Cells may also acquire drug-resistance via mutation, gene amplification, or epigenetic changes (Otto et al, 1989; see Chap. 19). Consequently, the rate at which drug-resistant variants arise in a cell population is an indirect measure of genetic instability. Using specific markers of drug resistance, it has been observed that gene amplification occurs at a high frequency in transformed cells (10–3 to 10–7 events per cell per generation), yet is almost undetectable in normal diploid fibroblasts (a frequency of less than 10–8; Tlsty, 1990).
If the amplified gene is a positive regulator of DNA replication or cell-cycle progression, gene overexpression may result in increased cellular proliferation leading to clonal selection and malignant transformation. Some amplified oncogenes, such as human epidermal growth receptor 2 (HER2) in breast cancer and the N-myc gene in neuroblastoma, are predictors of poor prognosis in those patients who harbor tumors containing them (see Chap. 7, Sec. 7.5 and Chap. 20, Sec. 20.3.3).
The structure and activity of chromatin can be altered by posttranslational modifications (eg, acetylation, phosphorylation, methylation, and ubiquitylation). Methylation of DNA is one of the main epigenetic modifications in humans and plays an important role in control of gene expression and normal cellular differentiation. It has been observed in more than half of the tumor-suppressor genes that cause familial cancers. In these tumors, there is methylation in normally unmethylated cytosine phosphate guanine (CpG) islands within the DNA. Methylation-induced transcriptional silencing begins early during the process of genetic instability and can affect many genes that are important in tumor progression. This includes methylation of genes involved in cell-cycle control (eg, p16INK4a; see Chap. 9, Sec. 9.2), transcription, hormone biology (ie, estrogen and progesterone receptor genes; see Chap. 20, Sec. 20.2), intracellular signal transduction (see Chap. 8), apoptosis (see Chap. 9, Sec. 9.4), DNA repair and tumor-suppressor genes (eg, retinoblastoma [Rb]; see Chap. 7, Sec. 7.6.4). Given that methylation is a potentially reversible state, this creates a target for novel therapeutic strategies involving gene reactivation. For example, both retinoic acid and 5′-aza-deoxycytidine can reverse DNA methylation and reactivate gene expression of normal regulatory genes (eg, cyclin-dependent kinases), thereby leading to regression of some human leukemias.
Histones are the core protein components of chromatin and their acetylation status regulates, in part, gene expression. Two groups of enzymes, the histone deacetylases (HDACs) and the acetyl transferases, determine the level of histone acetylation. Deacetylated histones are generally tightly coiled with the DNA and are associated with silencing of gene expression; in contrast, the acetylation of histones leads to the uncoiling of chromatin and is generally associated with gene expression. HDAC inhibitors promote acetylation and are emerging as a new class of potential anticancer agents for the treatment of solid and hematological malignancies. Examples of these agents include short-chain fatty acids (sodium butyrate, valproic acid), hydroxyaminic acids (SAHA [suberoylanilide hydroxamic acid], Trichostatin A [TSA]), synthetic benzamide derivatives, and cyclic tetrapeptides (see Chap. 19, Sec. 19.2). Depending on the cell type, inhibition of HDACs in cancer cells can lead to transcriptional activation of approximately 2% of human genes, including tumor-suppressor genes. Treating cells with HDAC inhibitors increases cell-cycle arrest, induction of apoptosis, and differentiation in cancer cells, in vitro and in vivo, by promoting p21WAF cdk-mediated cell cycle inhibition and downregulating proproliferative Raf/Mek/Erk cell signaling pathways (see Chap. 8, Sec. 8.2). Several HDAC inhibitors have shown antitumor activity in vivo at nanomolar concentrations and are being evaluated in phase I-II clinical trials alone, or in combination with demethylating agents (Momparler, 2003).
Normal tissues may have different inherent abilities to maintain their genetic integrity when faced with similar exogenous or endogenous damaging stimuli. Comparison of primary stromal versus primary epithelial cell cultures suggests that normal epithelial cells may be predisposed to genetic instability. Stromal cells (eg, fibroblasts) eventually lose the ability to proliferate in culture (termed cellular senescence) as a result of loss of chromosomal telomere DNA (see Sec. 5.6). Other cell types, however, such as human mammary epithelial cultures (HMECs), exhibit genetic alterations caused by sequential loss of DNA repair and checkpoint control during cell proliferation that enable an increased number of population doublings; rare cells can then escape cellular senescence and become immortalized (Tlsty, 1990; Fig. 5–2). There are also differences between stromal and epithelial cultures in their ability to initiate cell-cycle checkpoints following DNA damage. These and other observations may account for the higher incidence of epithelial-based compared to stromal-based tumors in humans. Importantly, recent studies have suggested that genetic stability is maintained in stem cells when compared to somatic cells, in part as a result of altered DNA repair pathways (Woodward and Bristow, 2009).
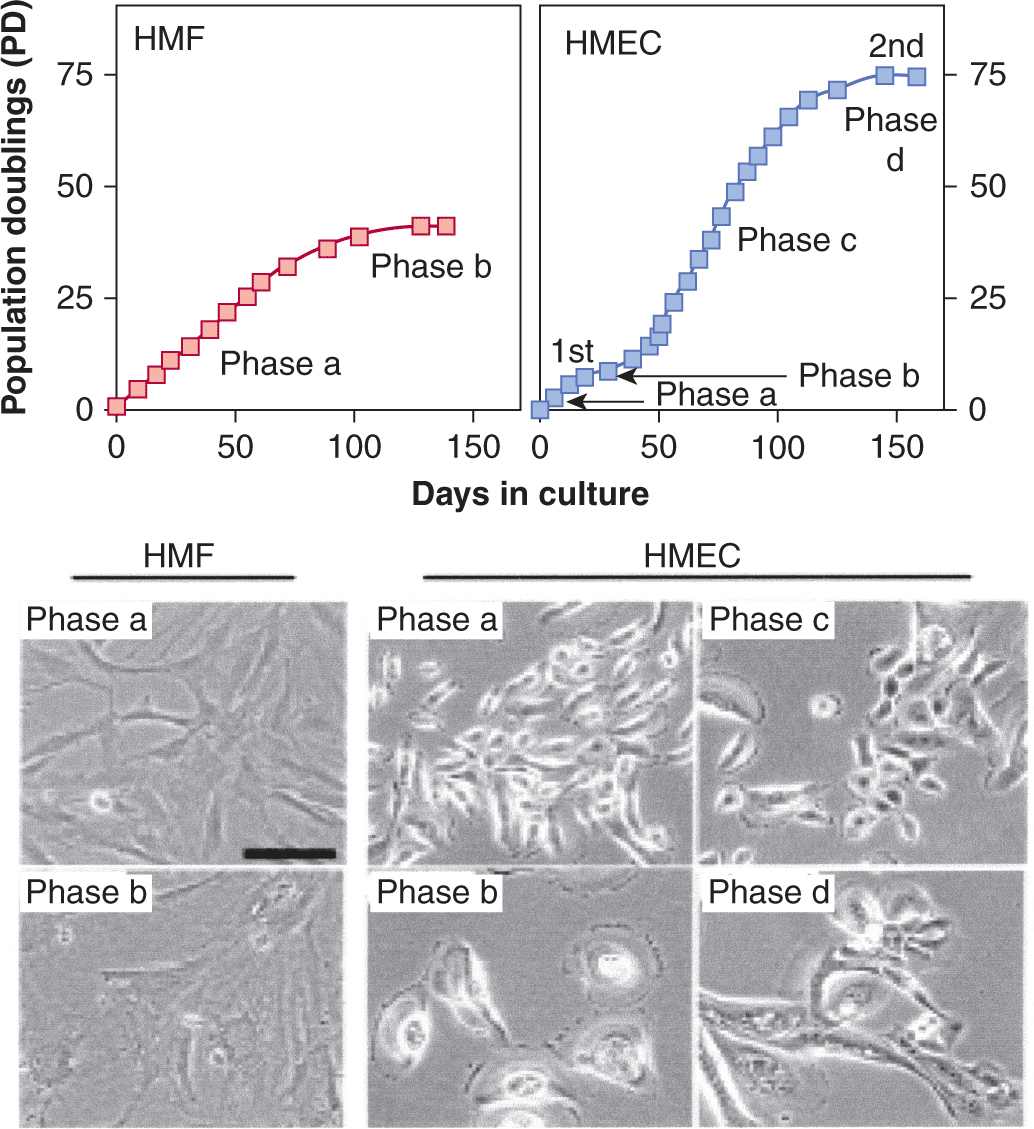
FIGURE 5–2 Genetic instability in epithelial tissues compared to stromal tissues. Data showing that human mammary fibroblasts (HMFs; left upper panel) undergo a limited number of cell divisions (phase a) before undergoing irreversible arrest, called senescence (phase b). In contrast, HMECs (upper right panel) exhibit an initial growth phase (phase a) that is followed by a transient growth plateau (termed selection or phase b), from which proliferative cells emerge to undergo further population doublings (phase c; approximately 20 to 70 doublings) before entering a second growth plateau (phase d). Seen in the panels below are representative cell images from each phase for each cell type. HMECs emerge from senescence, exhibit eroding telomeric sequences (see Sec. 5.6) and, ultimately, enter telomere-based crisis to generate the types of chromosomal abnormalities seen in the earliest lesions of breast cancer and point to differences between epithelial cells and fibroblasts during neoplastic transformation. (Data from Romanov et al, 2001.)
Multiple studies are attempting to document specific bio-markers of genetic instability in premalignant human tissues. This information can be used to ascertain whether an individual requires treatment with chemopreventive agents based on the biomarker panel, familial susceptibility, and carcinogenic insult. Tissue biomarkers can be related to mechanisms of DNA repair, cell-cycle checkpoint control, altered oncogenes or tumor-suppressor genes, or chromosomal aberrations. For example, biopsies have been obtained from tissues in high-risk patients (ie, bronchial biopsies from chronic smokers, oral or laryngeal biopsies from individuals with premalignancy) and examined for chromosome instability using in situ hybridization (Hittelman, 2001). Nearly all biopsy specimens show evidence for chromosome instability throughout the exposed tissue, often with multifocal clonal outgrowths that can persist for many years, perhaps accounting for continued risk of lung cancer following smoking cessation. Future investigations may lead to the discovery of new compounds that can reverse genetic instability in a given tissue, thereby preventing cancer.
5.2.2 Extrinsic Causes of Genetic Instability
In addition to reactions that occur spontaneously between DNA and the intercellular environment, many extracellular factors also place pressure on the genome. These extrinsic causes of genetic instability are produced by a myriad of sources including ultraviolet light (UV), ionizing radiation (IR), and chemical carcinogens (see Chap. 4).
5.2.2.1 Ultraviolet Radiation There is a correlation between latitude (average sun exposure) and the incidence of malignant tumors of the skin, with the tumors tending to occur on sun-exposed areas, such as the face. Genetic background is also a determining factor, especially low skin pigmentation, as this contributes to an increase in the effective dose delivered to the cells at risk in the basal layer of the epidermis. Chronic exposure to sunlight is required for carcinogenesis, suggesting the need for multiple interactions of UV radiation with the target cells.
UV-induced tumors in mice demonstrate point mutations in the TP53 tumor-suppressor gene (see Chap. 7, Sec. 7.6.1) and these mutations are primarily C-to-T transitions (Kress et al, 1992). More than 50% of skin cancers in humans (both squamous and basal cell carcinomas) also have characteristic p53 mutations (Ziegler et al, 1993). Many of these mutations are CC-to-TT transitions and are characteristic of misrepair or lack of repair of pyrimidine dimers in the DNA induced by the exposure to the UV radiation (Daya-Grosjean et al, 1995). Such dimers may be repaired by a number of processes, including nucleotide excision repair (see Sec. 5.3.3) and there is an increased risk of skin cancer in patients with xeroderma pigmentosum (XP), an inherited disease in which there is a deficiency in nucleotide excision repair (de Gruijl et al, 2001). The high incidence of p53 mutations found in preneoplastic lesions may cause genomic instability, which is consistent with the frequent loss of heterozygosity (LOH) seen in basal cell carcinoma (BCC; 9q) and squamous cell carcinoma (SCC; 3p, 9q, 13p, 17p, 17q), although LOH has also been observed in the absence of p53 mutations. The LOH at 9q also appears to be associated with deletion or mutation of the patched (PTCH) gene (see Chap. 8, Sec. 8.4.3) and 70% to 90% of BCC in XP patients have PTCH mutations (Daya-Grosjean and Sarasin, 2000). The PTCH gene is part of the Hedgehog (Hh) signaling pathway that can cause activation of the Gli transcription factors in human cells. One of the downstream targets of Gli is the Bcl2 gene, which acts to inhibit apoptosis (see Chap. 9, Sec. 9.4). Activation of this pathway may also override the G1-arrest associated with the p21 (WAF1) gene (see Chap. 9, Sec. 9.3 and Sec. 5.4).
In familial cutaneous melanoma there are markers on 9p21 that map to the INK4a,b locus, which codes for the cyclin-dependent kinase inhibitors p16 and p15 (see Chap. 9, Sec. 9.2.2). This locus also codes for the p14ARF gene, which can stabilize p53 by binding to the HDM2 gene and interfering with degradation of p53 (see Chap. 7, Sec. 7.6.1). Loss of INK4a (p16) appears to be the most important defect in familial cutaneous melanoma, while, in sporadic disease, point mutations typical of UV irradiation can be observed in this gene. Additionally, up to 70% of melanomas exhibit BRAF mutations that are associated with UV-induced lesions (Besaratinia and Pfeifer, 2008). Thus skin cancers are clearly associated with UV damage to DNA (Cleaver and Crowley, 2002; de Gruijl et al, 2001).
5.2.2.2 Ionizing Radiation The carcinogenic risks of radiation exposure have been derived from many sources, including occupational exposures (eg, radiologists and uranium miners), therapeutic exposures (eg, unavoidable treatment of normal tissues in cancer therapy, or treatment of ankylosing spondylitis), and accidental exposures (see Chap. 16, Sec. 16.8). Most information comes from studies of the A-bomb survivors in Hiroshima and Nagasaki, who were exposed in 1945, and from studies of exposures during medical x-ray examinations, particularly of pregnant women, which resulted in fetal exposure to irradiation. These groups of people were exposed to acute doses of radiation, and extrapolation of the risks from low levels of continuous exposure has relied on more limited information from occupational exposures and on experimental studies and modeling. Studies of radiation transformation in cultured cells point toward a high-frequency initial step followed by a rare second step (Little, 2000). The important initial effect of radiation appears to be the induction of genetic instability (Morgan et al, 2002; Syljuasen et al, 2001), which then allows for a higher probability of “rare” mutations that lead to malignant transformation. The observations, that radiation tends to increase the incidence of the types of tumors that arise naturally in the population and that exposure at earlier ages leads to increased relative risk, are consistent with the concept that radiation acts to induce genetic instability.
The mechanism(s) by which genetic instability is induced by radiation and maintained in the population are uncertain, but probably include (a) mutations in genes involved in control of DNA synthesis or DNA repair, such as the mismatch repair system (see Sec. 5.3.1); (b) the induction of chromosome instability; and (c) persisting aberrant production of oxygen radicals that can damage DNA (Little, 2000; Morgan et al, 2002). Irradiated cells maintain a higher incidence of mutations and chromosome instability for many generations after the exposure both in vitro and in vivo (Morgan et al, 2002). These unstable cells may continually acquire further lesions such as point mutations, that may produce mutator phenotypes.
Hanahan and Weinberg (2010) proposed a model where the induction of genetic instability is a key driver of the subsequent mutagenic events that induce malignant transformation. Ionizing radiation induces deletions, translocations, or inversion of DNA sequences that may induce genetic instability secondary to DNA repair and cell-cycle checkpoint defects (Cox, 1994). Alternatively, chromosome breakage followed by faulty repair and translocation or amplification of DNA segments are also possible mechanisms for activation of genetic instability.
5.3 DNA REPAIR PATHWAYS
Recall that on the order of 105 DNA lesions are produced per cell per day (Sec. 5.2.1). Cells have evolved multiple mechanisms to detect and remove these lesions to prevent propagation to progenitor cells and, in the case of multicellular organisms, suppress tumorigenesis. An improved understanding of the mechanisms of DNA repair came from the isolation of repair-deficient rodent cells (ie, Chinese hamster ovary [CHO] mutants) with unusual sensitivity to different classes of DNA-damaging agents. Some mutants exhibited extreme sensitivity to UV light and crosslinking agents, such as mitomycin C, but little or no sensitivity to x-rays. Other cells exhibited sensitivity to x-rays and chemical agents known to cause DNA breakage, but little or no sensitivity to UV light or crosslinking agents. These various phenotypes, which are similar to those characterized previously in bacteria and yeast, indicate the involvement of several distinct DNA repair pathways and associated gene products. Some of these repair pathways are so highly conserved from yeast to humans that yeast proteins can substitute for human proteins and vice versa; this has been helpful in the cloning and functional characterization of the human homologs of yeast DNA repair genes.
Some frequent lesions, such as those formed by oxidation or DNA-reactive carcinogens, induce structurally distinct mutagenic and cytotoxic damage to the DNA (see Chap. 4, Sec. 4.3). Some of these adducts are recognized and repaired by a class of enzymes that are used only once. For example, induction of O(6)-methylguanine (O(6)-MeGua) by oxidation or N-nitroso compounds is recognized by O(6)-alkylguanine DNA alkyltransferase, which reverts the O(6)-MeGua back to guanine in a single-step irreversible reaction that inactivates the enzyme and prevents mutagenic G:C→A:T transitions.
An important property in DNA repair is the fidelity of the repair pathway leading to the concepts of error-prone, and error-resistant (or error-free), DNA repair. Many DNA lesions can block transcription of RNA, thereby inactivating the DNA damage-containing gene on the DNA strand that is being transcribed. Persistent blockage of RNA synthesis can lead to cell death so these lesions are often repaired through the transcription-coupled repair pathway (see Sec. 5.3.3 below); this pathway is designed to displace the stalled RNA polymerase and drive a high-priority repair mechanism. For lesions that block progression of the replication fork during DNA replication, several error-prone DNA polymerases have been described that have increased flexibility and low fidelity to allow for replicative bypass (ie, translesion DNA synthesis) of the base damage contained within DNA. These polymer-ases can be used temporarily by the cell during acute DNA replication damage and substituted by more accurate DNA polymerases at a later time. Use of these lower-fidelity bypass DNA polymerases can contribute to high error-rates during DNA replication and may lead to malignant transformation. This process is diagrammed in Figure 5–3 and is known as translesion DNA synthesis.
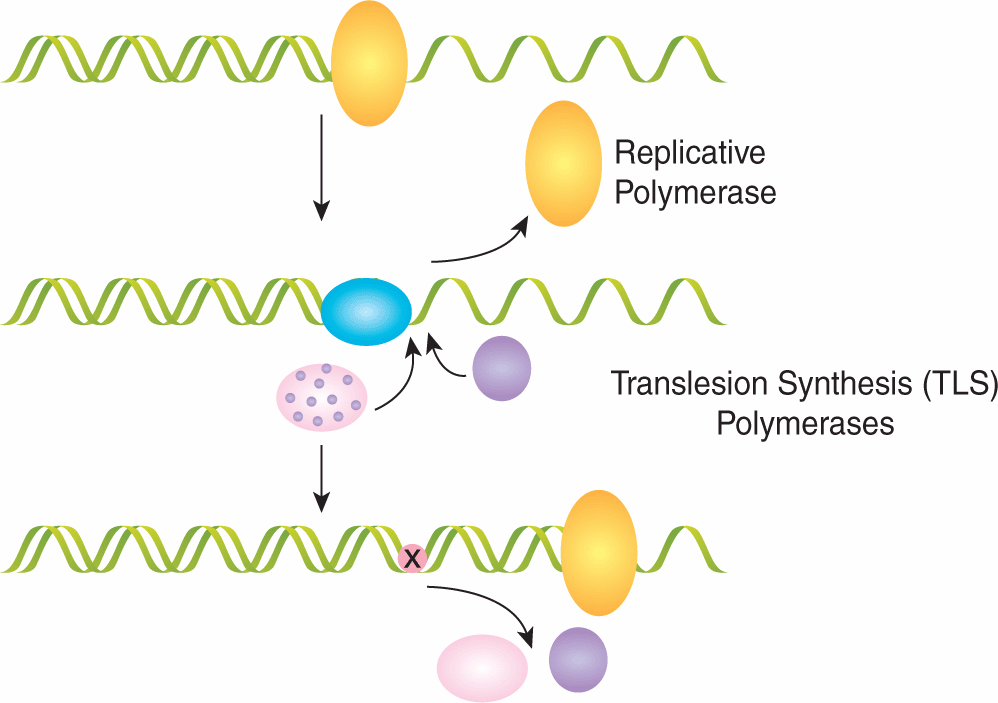
FIGURE 5–3 Translesion DNA synthesis. The “DNA polymerase switch model” for translesion synthesis and mutagenesis. This cartoon shows the replicative DNA polymerase (yellow sphere) blocked at a template lesion site. Cells contain several different DNA polymerases (other indicated spheres) that transiently replace the replicative DNA polymerase. After a short patch of synthesis in the vicinity of the lesion, the replicative DNA polymerase resumes high fidelity and processive synthesis. (Adapted from Cordonnier and Fuchs, 1999.)
In the following sections, discrete biochemical pathways of DNA repair are described. These pathways can be divided into different classes depending on the specific DNA lesion they are designed to repair, and include (a) mismatch repair, (b) base excision repair, (c) nucleotide excision repair, (d) singlestrand break repair, and (e) homologous and nonhomologous repair of DNA double-strand breaks.
5.3.1 Mismatch Repair
The mismatch repair (MMR) pathway is enacted when the DNA-polymerase inserts an incorrect base during DNA replication or when the polymerase creates helical distortions by inserting or deleting bases in short oligonucleotide repeats (microsatellites) during replication. These helical distortions are termed insertion–deletion loops. The protein products of MMR genes form heterodimer complexes, and different protein pairs recognize specific mismatched nucleotides or insertion-deletion loops in DNA (Fig. 5–4). For example, the MSH2 protein forms a heterodimer with an additional MMR protein, MSH6 or MSH3, and the resulting complexes are called MutS-α or MutS-β, respectively. MUTS-α is required for the recognition of DNA base–base mismatches, whereas MutS-α and MutS-β have partially redundant functions for the recognition of DNA insertion–deletion loops. A second heterodimer forms between the MMR gene product MLH1 and PMS2 or MLH3 to form MutLα and MutLβ, respectively. The MutL complexes coordinate the interplay between the initial mismatch recognition complex and subsequent protein interactions required to complete MMR. The latter proteins include proliferating cell nuclear antigen (PCNA), DNA polymerases γ and ε, and possibly DNA helicases that unwind the DNA helix to facilitate DNA synthesis (Hoeijmakers, 2001). These processes are diagrammed in Figure 5–4.
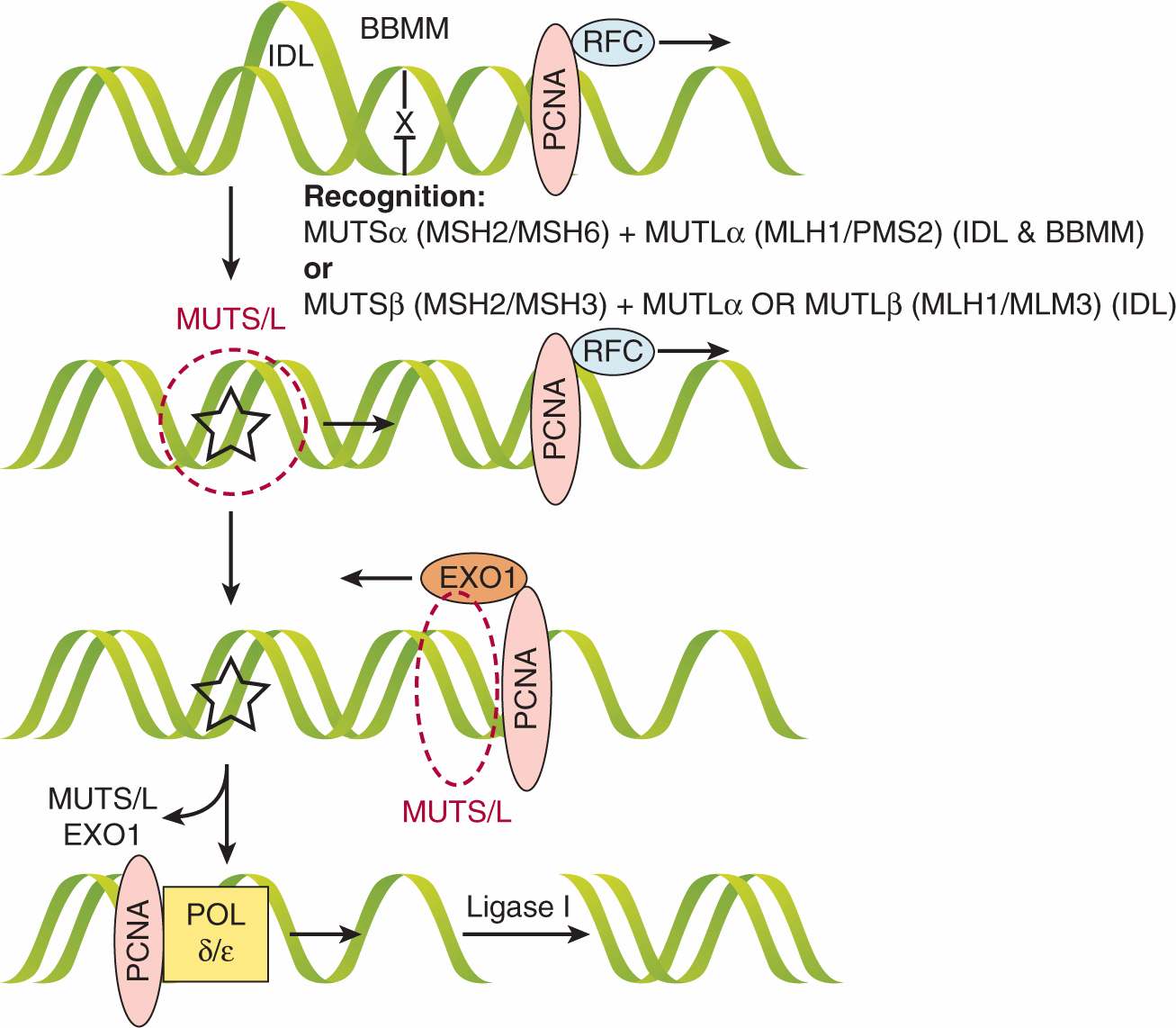
FIGURE 5–4 Mismatch repair (MMR). The mismatch repair pathway is initiated either when a base is misincorporated to create a base–base mismatch (BBMM) or when insertion–deletion loops (IDLs) are created during replication in microsatellite regions. These lesions are recognized by different complexes: MSH2/MSH6 (MutSα) + MLH1/PMS2 (MutLα) recognize BBMMs and IDLs, whereas MSH2/MSH3 (MutSβ) + MutLa or MLH1/MLH3 (MutLβ) can also recognize IDLs. Whichever MutS/L complex is utilized, it translocates along DNA until it reaches the proliferating cell nuclear antigen (PCNA) associated with the replication factor complex (RFC) at the site of the replication fork. Next, the nuclease EXO1 is loaded and activated, which resects the fork backward past the lesion, at which point MutS/L and EXO1 are unloaded. The DNA polymerase δ and ε(POL δ/ε) fills the gap and Ligase I seals the nick if necessary. (Adapted from Jiricny, 2006.)
5.3.2 Base Excision Repair
Spontaneous oxidative damage is known to occur in cells producing 104 to 105 oxidative residues, such as 8-oxodeoxyguanosine, per cell per day among the approximately 3 × 109 bases in the genome. DNA base damage, occurring as a result of endogenous oxidative processes or exogenous DNA damage (eg, from ionizing radiation) is repaired by the base excision repair pathway. Base excision repair involves the enzymatic removal of the damaged DNA base by DNA glycosylases. DNA glycosylases are a family of enzymes that cleave glycosidic bonds and are specific to particular base lesions. There are 2 classes of DNA glycosylases that differ in their reaction mechanism: monofunctional enzymes leave the DNA strand intact and bifunctional DNA glycosylases also cleave the DNA backbone (Fig. 5–5). For example, the OGG1 protein is a bifunctional 8-oxoguanine DNA glycosylase that removes spontaneous or ionizing radiation-induced lesions to prevent cellular mutations. During base excision repair the initial base removal step leaves an apurinic or apyrimidinic site that is similar to single-strand DNA breaks. Such single-strand breaks can be induced by free radicals or by ionizing radiation without the action of DNA glycosylases, and repair of these 2 types of lesions (base damage and single-strand breaks) converge into this common pathway. Base excision repair and single-strand break repair involve similar components and the processes are shown in Figure 5–5. The major pathway is short-patch base excision repair and involves the replacement of a single nucleotide following DNA backbone cleavage at the base excision site. A minor pathway is the long-patch base excision-repair pathway, which exists for the repair of 2 to 13 damaged nucleotides.
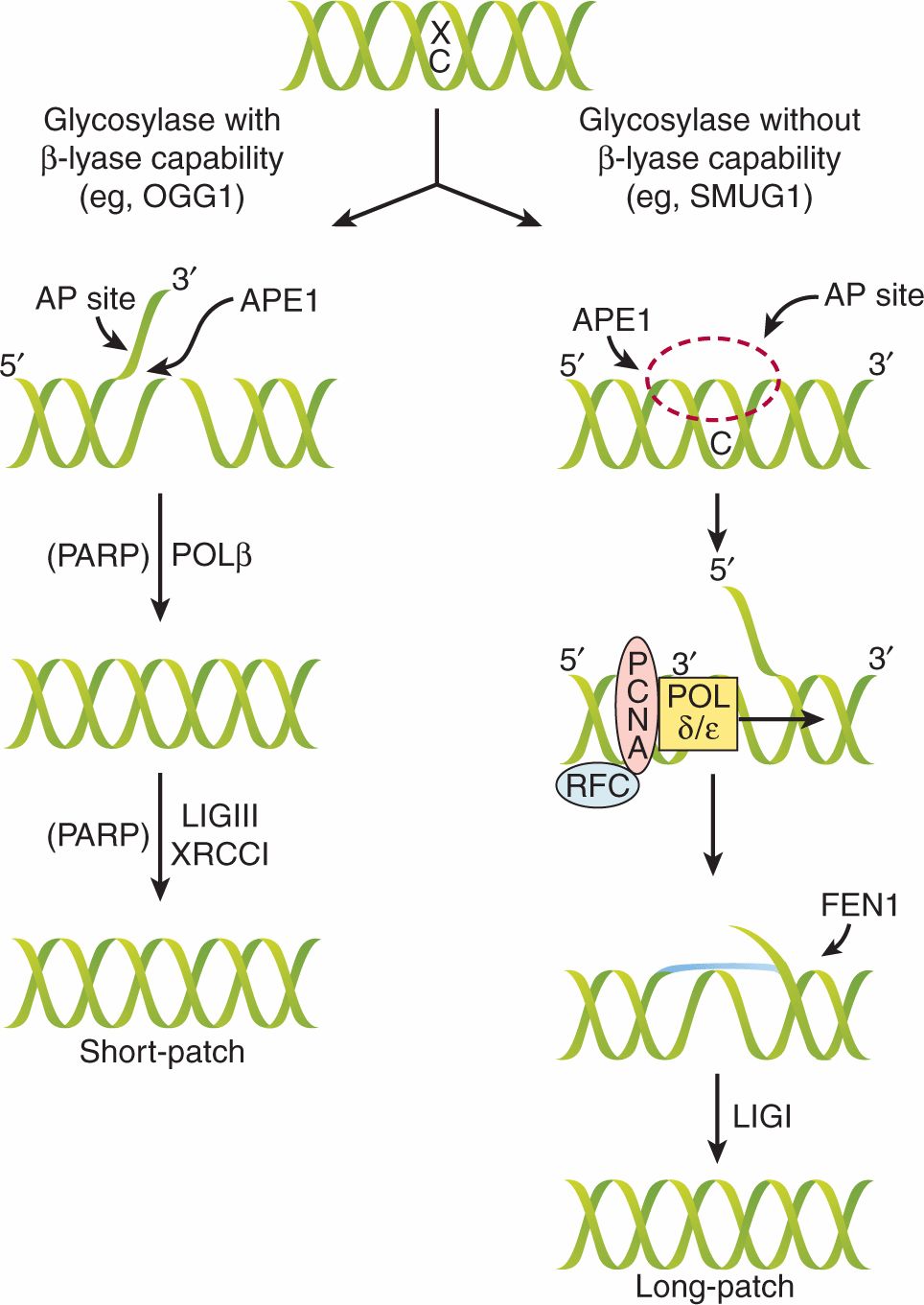
FIGURE 5–5 Mechanism of base excision repair (BER) and single-strand break repair (SSBR). Damaged bases are removed by the BER pathway and single-strand breaks are repaired by components of the same pathway. There are 2 subpathways, known as short- and long-patch BER, and pathway choice depends on the initial glycosylase enzyme that is required to recognize and remove the particular base lesion. When the enzyme includes β-lyase activity, the base is removed, leaving an apyrimidinic (AP)-site with a nick in the DNA backbone and a flap 3′ to the removed base. The APE1 endonuclease removes this flap and the POLβ enzyme fills in the gap. Simultaneously the single-stranded DNA recruits the poly(ADP) ribose polymerase (PARP) enzyme that facilitates recruitment of ligase III and XRCC1 that seal the nick. Because only a single nucleotide is replaced, this is called short-patch BER. In the complementary long-patch BER pathway the recognition glycosylase does not contain β-lyase activity and leaves an AP site with an intact DNA backbone. The APE1 endonuclease catalyses the formation of a flap 5′ to the removed base, and PCNA, POLδ/ε, and replication factor C (RFC) initiate synthesis and create a longer flap of 2 to 10 nucleotides. The FEN1 nuclease cleaves the overhang and the nick is sealed by ligase I. (Adapted from Sancar et al, 2004.)
No human disorders have been related directly to inherited deficiencies in base excision repair, and knockout mice engineered to lack core proteins in the pathway die as embryos, attesting to its important role in development. Genetic mouse knockout models for a variety of glycosylase genes have shown only mild increases in genetic mutations. This may be because of partial redundancy in the glycosylases and/or overlap with the transcription-coupled repair processes described below. However, base excision repair may be defective in cells that have mutations in p53 as the p53 protein can stimulate base excision repair by direct interactions with APE1 and DNAPolβ (Offer et al, 2001). Indeed, the gene locus encoding the glycosylase 8-Oxoguanine glycosylase (OGG1) on chromosome 3p25-26 is frequently lost in lung cancers, consistent with a purported role in preventing carcinogenesis.
An exciting area of exploration is the clinical utility of poly(ADP)-ribose polymerase (PARP) inhibitors. As shown in Figure 5–5, PARP activity is required as an intermediate step preceding DNA synthesis during base excision repair or when single-stranded DNA regions are recognized. When PARP is inhibited by small molecule inhibitors, this causes the accumulation of single-stranded gaps that can cause collapse of replication forks during DNA replication. Under normal circumstances homologous recombination (see below) during DNA replication can rescue this and prevent double-strand break (DSB) formation. However, when homologous recombination is defective, such as in BRCA2-deficient breast cancers, these PARP inhibitors can lead to accumulation of DSBs and cell death (Helleday, 2010). This general concept of inhibiting compensatory pathways in tumor cells, while sparing normal tissues, is termed synthetic lethality, a common concept in yeast biology, and it has become an exciting area of research for novel cancer therapies (see Chap. 17, Sec. 17.3.2).
5.3.3 Nucleotide Excision Repair
In aqueous solution, DNA is susceptible to absorption of photons in the range of 200 to 300 nm, which increases reactivity of pyrimidine bases to produce 6-4 photoproducts (6-4PPs) and interstrand crosslinks in the form of cyclobutane pyrimidine dimers (CPDs; eg, thymine–thymine linkages). These lesions, and other bulky chemical adducts in DNA, are removed by nucleotide excision repair (NER), which is a complex DNA repair pathway involving more than 30 genes. Many of the NER genes were originally cloned from complementation analyses of cells from patients with XP and with cells from patients with Cockayne syndrome (CS), and are referred to as XPA-XPG or CSA or CSB in protein nomenclature. Patients with these syndromes have severe sensitivity to UV light and a much higher incidence of skin cancer (Fig. 5–6).
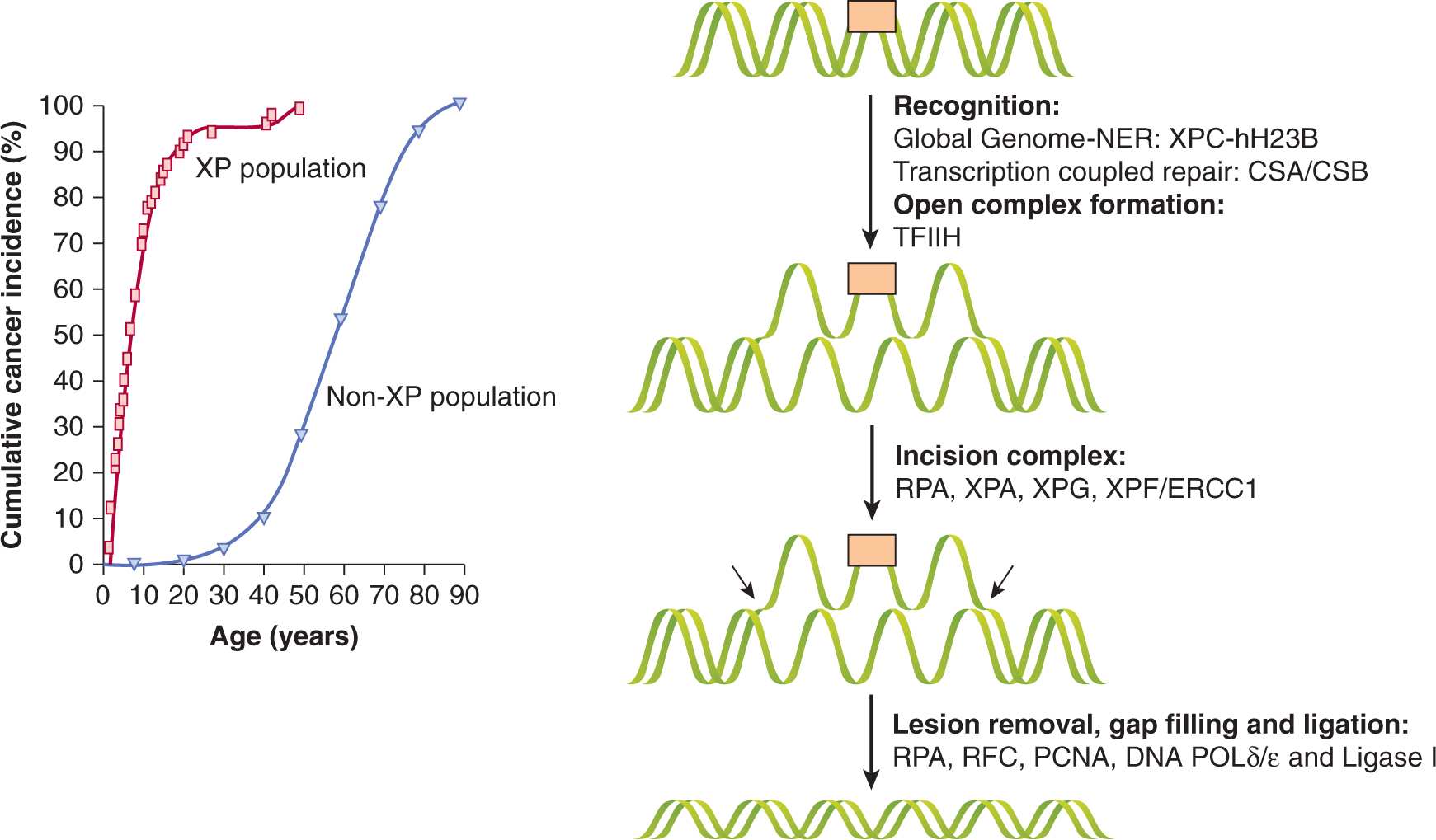
FIGURE 5–6 NER and XP. Left: Incidence of skin cancer in people with XP compared to the normal population with similar exposure to the sun. The 1000-fold excess risk of UV-induced skin cancer is secondary to defective repair of UV-induced DNA lesions in XP patients. Right: NER proceeds by initial recognition of the lesion by 1 of 2 different complexes, depending on whether “global genome”-nucleotide excision repair (GG-NER) or “transcription-coupled” repair (TCR) is used. In GG-NER helical distortions stimulate recognition by the xeroderma pigmentosum complementation group C (XPC)-hHR23B complex. Subsequently, XPA is recruited and transcription factor IIH (TFIIH) causes formation of an “open complex.” The single-stranded regions of DNA are recognized by RPA. Two endonucleases, XPG and XPF/ERCC1, cut 3′ and 5′ to the lesion, respectively. The approximately 30-nucleotide gap is filled by POLγ/ε and the nick is sealed by ligase I. In TCR, the RNA polymerase II transcription machinery is thought to facilitate recognition of the lesion that, along with the CS proteins A and B (CSA/B), contribute to recognition of the lesion and induction of NER in a similar mechanism to GG-NER. The entire process of NER takes several minutes to complete.
The process of NER is highly conserved in eukaryotes and consists of the following 4 steps: (a) recognition of the damaged DNA; (b) excision of an oligonucleotide of 24 to 32 residues containing the damaged DNA by dual incision of the damaged strand on each side of the lesion; (c) filling in of the resulting gap by DNA polymerase; and (d) ligation of the nick (Balajee and Bohr, 2000). In human cells, NER requires at least 6 core protein complexes for recognition of damage and dual incision (XPA, XPC-hHR23B (human homolog of RAD23B), RPA (replication protein A), TFIIH (transcription factor IIH), XPG and ERCC1-XPF (excision repair cross complementation 1) and other factors for DNA synthesis and ligation to complete repair (PCNA, RFC [replication factor C], DNA polymerase δ or α, and DNA ligase I) (de Laat et al, 1999). The process of NER is diagrammed in Figure 5–6.
NER consists of 2 subpathways that differ in their mode of recognition of the helical distortions that the lesions produce. The first subpathway, termed global genome repair (GG-NER) is transcription-independent and surveys the entire genome for DNA lesions. The 6-4PPs, which distort the DNA more than CPDs, are removed rapidly, through recognition by the XPC-HH23B protein complex in GG-NER. In contrast, CPDs are repaired very slowly by GG-NER and are removed more efficiently from the transcribed strand of expressed genes by transcription-coupled repair (TCR). During TCR, the stalled RNA polymerase induces the recognition of the DNA lesions on the transcribed strand, and this process is facilitated by CSA and CSB proteins (Friedberg, 2001). The TCR and GG-NER pathways converge after this recognition step to form the open complex and removal of the damage as shown in Figure 5–6.
5.3.4 DNA Double-Strand Break Repair: Homologous Recombination
DSBs in DNA result from ionizing radiation and certain chemotherapeutic drugs, from endogenously generated reactive oxygen species, and from mechanical stress on the chromosomes (Zhou et al, 1998). They can also be produced when DNA replication forks encounter DNA single-strand breaks, following defective replication of chromosome ends (ie, telomeres, see Sec. 5.6) or when topoisomerase enzymes are inhibited (eg, by etoposide) preventing the rejoining of the DSBs these enzymes induce. In addition, DNA DSBs are generated to initiate recombination between homologous chromosomes during meiosis and occur as intermediates during developmentally-regulated rearrangements, such as V(D) J recombination during the generation of immunoglobulins (see Chap. 21, Sec. 21.3.1).
In human cells, repair of DNA DSBs occurs either by homologous recombination (HR; Fig. 5–7) or nonhomologous end-joining (NHEJ; Fig. 5–8). The preferred pathway depends on tissue type, the extent of DNA damage, the cell-cycle phase in which the cell is damaged, and the relative need for repair fidelity. There may also be cooperation between the 2 pathways (Richardson and Jasin, 2000). Repair by HR requires homology between the broken DNA strand and the template strand used in repair. Typically, this is newly replicated sister chromatid and as a result HR is restricted to the S and G2 cell-cycle phases (see Fig. 5–7). The HR pathway results in error-resistant repair of DNA DSBs because the intact undamaged template is used to pair new DNA bases between the damaged and undamaged strands during DNA synthesis.
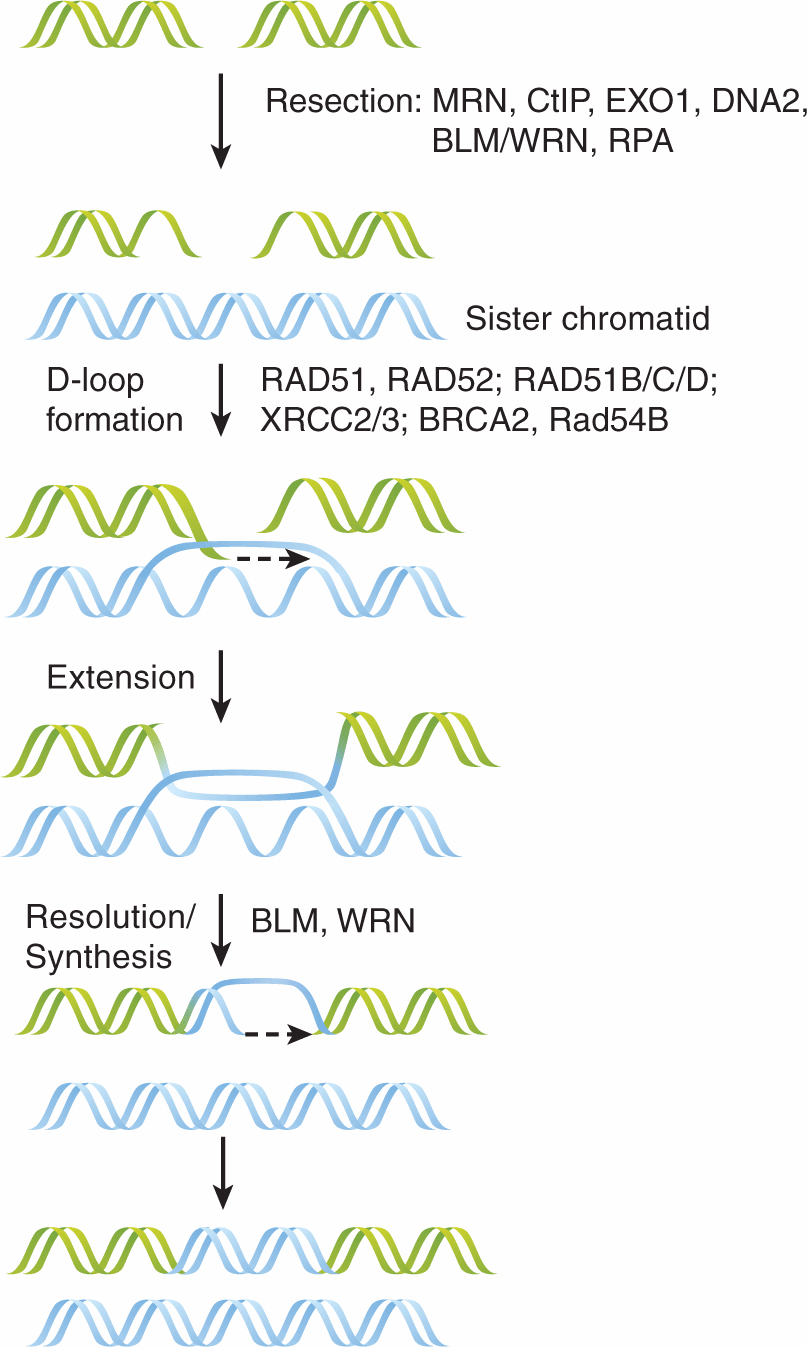
FIGURE 5–7 HR of DNA DSBs. HR is a major pathway for DSB repair and requires the presence of a sister chromatid. Here, single-stranded DNA overhangs are created and stabilized by nucleases (MRN [MRE11-RAD50-NBS1] complex, CtIP, EXOI, DNA2), helicases (BLM, WRN), and the single-strand DNA binding protein RPA. These RPA-coated overhangs are then exchanged to create RAD51-coated overhangs, a process facilitated by the RAD52 group of proteins (RAD52, RAD51B/C/D, XRCC2/3), BRCA2, and RAD54B, that also contributes to scanning for homologous DNA regions. This RAD51-filament can then invade the complementary DNA-duplex to form a D-loop. DNA polymerases then extend this invading strand and the corresponding strand on the opposite side of the DSB. (Adapted from Helleday et al, 2007.)
The HR pathway is highly conserved, likely owing to its ability to maintain the integrity of genetic information. Shown in Figure 5–7 is a general schematic of HR processes where a DSB is initially recognized by the MRN complex (MRE11-RAD50-NBS1). MRE11 possesses both single-stranded DNA (ssDNA) endonuclease and 3′ to 5′ exonuclease activities, which are enhanced by CtIP. Together with the 5′ to 3′ exonuclease EXO1 and endonuclease DNA2, these proteins collaborate with heli-cases (ie, BLM and WRN) to form resected ssDNA, which is rapidly coated with replication protein A (RPA). Exchange of RPA for the RAD51 protein is facilitated by the RAD52 epistasis group, XRCC2/3, BRCA1/2, and RAD54B, which also contributes to strand invasion of the sister chromatid forming a D-loop (Heyer et al, 2010; Svendsen and Harper, 2010). DNA synthesis extends the invading strand and the D-loop is resolved by poorly understood mechanisms. There are actually several subpathways of HR that vary in their ability to prevent crossover of information from the strand acting as the template for DNA synthesis during the formation of so-called Holliday junctions (see Fig. 5–7; Helleday et al, 2007; Heyer et al, 2010).
The BRCA1/2 breast cancer-susceptibility proteins (see also Chap. 7, Sec. 7.6.3) also play a role in the homologous repair of DNA DSBs. Both BRCA1 and BRCA2 proteins form discrete nuclear foci during S-phase following exposure to DNA damaging agents at the sites of DNA damage. Although RAD51 colocalizes at subnuclear sites with BRCA1, their interaction is thought to be indirect, with only 1% to 5% of BRCA1 in somatic cells associating with RAD51 (Marmorstein et al, 1998). In contrast, the BRCA2 protein contains 8 BRC repeats, each of 30 to 40 residues, which are the major sites for the direct binding to RAD51 by a substantial fraction of the total intracellular pool of BRCA2 (Davies et al, 2001). As such, BRCA2-deficient cells have 10-fold lower levels of HR when compared to BRCA2-proficient cells (Moynahan et al, 2001). One model suggests that a BRCA2-RAD51 complex promotes the accurate assembly of DNA repair proteins required to offset DNA breaks that accumulate during DNA replication; these could otherwise lead to gross chromosomal rearrangements, LOH at tumor-suppressor gene loci, and carcinogen-esis. In some instances, when a single-stranded gap is met by the replication machinery, a single DSB will be produced, which utilizes HR as a mechanism to restart replication forks if the delay is not prolonged.
Biochemical and genetic studies in yeast have been fundamental in the ability to clone human homologs of proteins involved in the HR pathway. In the yeast, Saccharomyces cerevisiae, the RAD52 group of genes are involved in HR including RAD50, RAD51, RAD52, RAD54B, RAD55, RAD57, RAD59, MRE11, and XRS2 (the latter retermed p95 or NBS1-nibrin in mammalian cells). RAD51–/– mice are embryonic lethal, attesting to the importance of this critical HR protein in meiosis and development. Careful observations during the initial stages of embryogenesis in RAD51–/– mice show that lethality is preceded by chromosomal rearrangements and deletions. It is thought that DNA replication errors and replication-associated DNA strand breaks are converted into DNA DSBs in HR-defective cells (Lim and Hasty, 1996). In cells derived from RAD54–/– mice (which are developmentally normal), there is also decreased HR and increased hypersensitivity to DNA crosslinking agents such as mitomycin C (Essers et al, 1997). Although in S. cerevisiae RAD52 is essential for DNA DSB repair, RAD52–/– mice are viable and fertile and do not show a DNA DSB repair deficiency. HR therefore appears to be more complex in mammalian cells than in yeast, possibly as a result of functional redundancy in many of the proteins.
A number of human cancers, including ovarian, breast, prostate, and pancreatic, have mutations or altered expression and function of the MRE11, RAD51/RAD52/RAD54, and BRCA1/2 genes. This observation suggests that tumorigenesis is associated with altered HR in sporadic tumors. Increased levels of RAD51 expression in certain cancer cell lines also has been associated with altered phosphorylation, ubiquitination and transcription of the RAD51 protein as a result of abnormal c-ABL– and STAT5-mediated tyrosine kinase signaling pathways in cancer cell lines (see Chap. 8). This can lead to acquired radioresistance and chemoresistance (Daboussi et al, 2002).
5.3.5 DNA Double-Strand Break Repair: Nonhomologous End-Joining
The NHEJ pathway is outlined in Figure 5–8. The recognition step of NHEJ is initiated by high-affinity binding of the KU70/80 heterodimer to the DNA ends, which causes a conformational change that recruits DNA-dependent protein-kinase catalytic subunit (DNA-PKcs). Autophosphorylation of DNA-PKcs at multiple sites is essential for NHEJ to occur and appears to mediate DNA-PKcs dissociation from the break site (Dobbs et al, 2010). Many DNA DSBs have damaged ends that need to be processed to restore their ability to be ligated; for example, if 5′-phosphates are not present and/or 3′-phosphate groups need to be removed to create blunt ends. These structures are repaired by “end processors” that are partially dependent on the activity of the kinase mutated in ataxia telangiectasia (ATM [ataxia-telangiectasia mutated]) and include the Artemis protein, CtIP and MRN complexes, which are nucleases, PNKP which is both a 5′ kinase and 3′ -phosphatase and Polμ/λ. Once ends are restored the XRCC4-ligase IV complex, stimulated by XRCC4-like factor (XLF), rejoins the ends (Hiom, 2010). MMEJ and single-strand annealing (SSA), are minor subpathways of NHEJ that require end resection to reveal short (5 to 25) and longer (>30) homologous stretches of DNA, respectively (see Fig. 5–8). Although this pathway is much less understood, it is believed to contribute to deletions and chromosomal rearrangements that result in genomic instability (McVey and Lee, 2008).
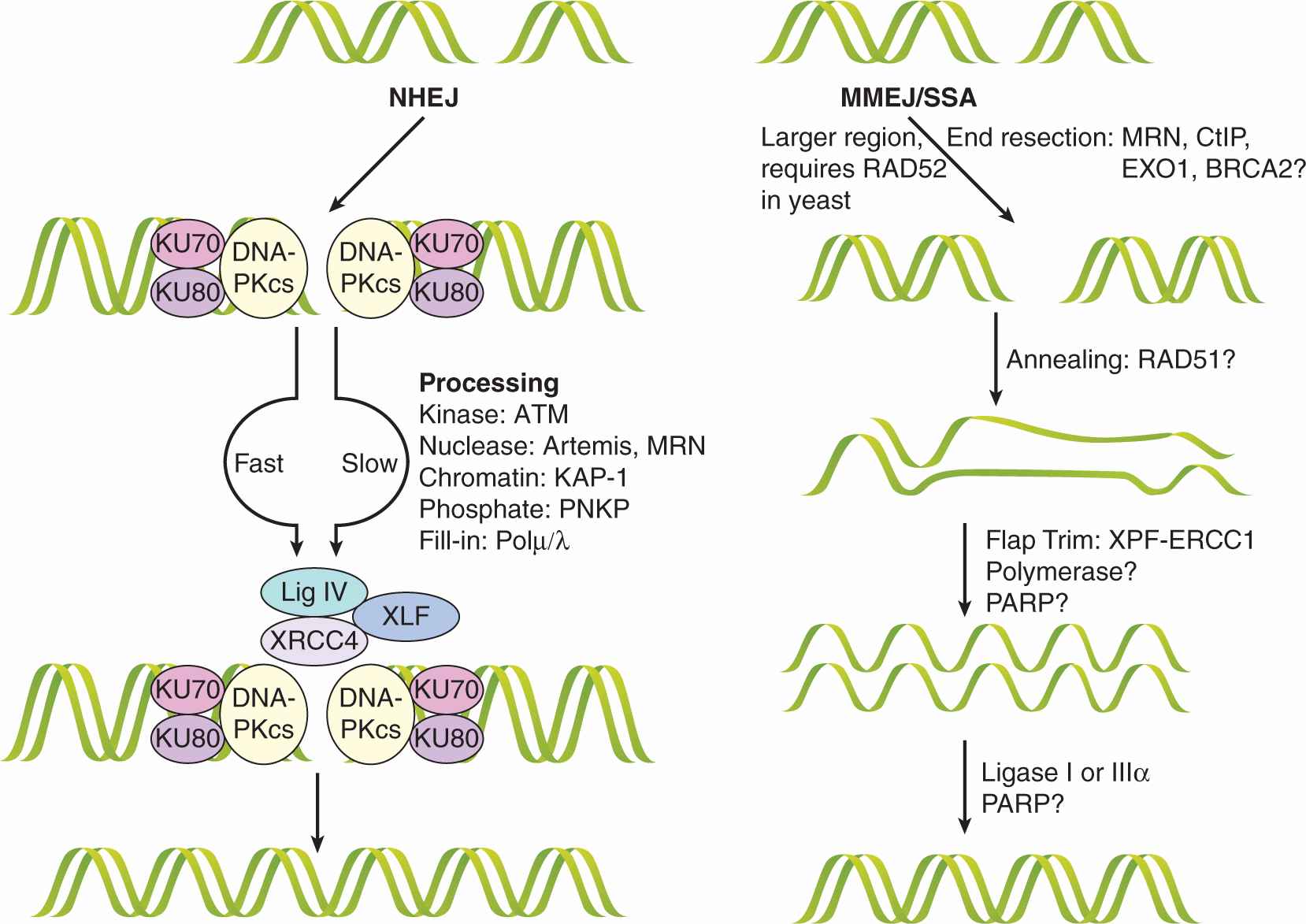
FIGURE 5–8 NHEJ of DNA DSBs. NHEJ is composed of subpathways that repair the DNA lesion. In the “fast” NHEJ pathway, the KU70/80 heterodimer recognizes the DNA ends and recruits the DNA-dependent protein-kinase catalytic subunit (DNA-PKcs), which autophosphorylates and forms the active DNA-PK complex. Downstream the ligase IV/XRCC4 dimer associates with the XLF/Cernuous proteins and joins the DNA ends together. In some cases, the DSB ends at the break are unligatable and require processing in the so-called slow NHEJ pathway. Repair of these ends depends at least in part on the ATM kinase and is processed by nucleases such as MRE11 or Artemis, kinases or phosphatases like PNKP, or filled in by Polμ/λ. Once competent for ligation, these ends follow the same XRCC4/ligase IV pathway above. Some lesions may require chromatin modification by KAP-1 for repair.
A less-well understood parallel pathway is microhomology-mediated end-joining (MMEJ)/single-strand annealing (SSA), which can be thought of as a hybrid pathway for DSB repair. The distinguishing features of MMEJ and SSA are poorly defined, but are differentiated primarily by the extent of the homologous regions involved and the requirement of RAD52 for SSA in yeast. The initial step includes end resection to expose homologous regions by nucleases, including MRE11, CtIP, EXO1, and BRCA2. The annealing of the homologous single-strand overhangs is facilitated by the RAD51 protein and the remaining flaps are trimmed by XPF-ERCC1. The involvement of polymerases and the PARP enzyme in this step is likely necessary but unknown. The nicks are sealed with ligase I or IIIα. Despite the disadvantage of its low fidelity, this pathway can act quickly, as required of an emergency mechanism, and, unlike HR, it does not depend on sister DNA molecules, which exist in the cells only after DNA replication. (Adapted from Dobbs et al, 2010; McVey and Lee, 2008.)
The major protein complexes implicated in the NHEJ pathway are the DNA-dependent protein kinase (DNA-PK) complex and the XRCC4/ligase IV complex. Human DNA-PK consists of an approximately 460-kDa DNA-PK catalytic subunit (DNA-PKcs), and a DNA end-binding KU heterodimer (consisting of 70-kDa and 80-kDa protein subunits). The catalytic subunit shows homology to the phophatidylinositol-3 kinase (PI3K) superfamily at its C-terminus, which contains the protein kinase domain required for phosphorylating DNA-PK–associated proteins during repair. Mutations in either DNA-PKcs or in one of the KU genes result in sensitivity to ionizing radiation and reduced ability to repair radiation-induced DNA DSBs. XRCC4 forms a stable complex with DNA ligase IV and XLF, and probably links detection of the initial lesion by DNA-PK to the actual ligation reaction carried out by ligase IV (see Fig. 5–8) (Pang et al, 1997).
Much information regarding the cellular activity of the DNA-PK complex stems from research utilizing the severe combined immunodeficiency (SCID) model mouse, which has a complete lack of mature T and B cells and is radiosensitive. The DNA-PKcs protein in the SCID mouse is mutant and unstable because of a loss of the last 83 amino acids prior to the C-terminal kinase domain. Consequently, DNA-PK activity is severely reduced in tissues derived from this animal. Immunodeficiency is secondary to an inability to process and rejoin the broken DNA molecules produced endogenously during rearrangement of immunoglobulins and T-cell receptor loci (see Chap. 21, Sec. 21.3.1). These animals also show chromosomal instability in their normal cells and are susceptible to lymphoma, suggesting that these act as tumor-suppressor genes (Khanna and Jackson, 2001). Consistent with phenotypes observed in the animals, fibroblasts derived from DNA-PKcs, KU70 or KU80 deficient mice present delayed kinetics of DSB repair and overall lower DSB rejoining following ionizing radiation (see Chap. 15).
The RAD50–MRE11–NBS1 protein complex (termed MRN) acts in both HR and NHEJ pathways (see Figs. 5–7 and 5–8) and also in maintenance of telomeres (see Sec. 5.6). Mutations in the NBS1 gene (also called the p95 or nibrin gene in humans) result in Nijmegen breakage syndrome (NBS), a recessive disorder with some phenotypic similarities to ataxia telangiectasia (AT); (see Sec. 5.5), including chromosomal instability, radiosensitivity, and an increased incidence of lymphoid tumors (Featherstone and Jackson, 1998; Little, 1994). Mutations in human MRE11 have been linked to the ataxia-telangiectasia–like disorder (ATLD). Cells from NBS, AT, and ATLD patients are hypersensitive to DSB-inducing agents and show radioresistant DNA synthesis (persistent DNA synthesis after irradiation that is not observed in normal cells) after exposure to ionizing radiation (Girard et al, 2000). Disruption of the mammalian RAD50 or MRE11 genes results in nonviable mice attesting to their importance in development. Biochemical studies of the yeast and human protein complexes have shown that MRE11 has a 3′ to 5′ Mn2+-dependent exonuclease activity on DNA substrates with blunt or 5′ protruding ends and endonuclease activity on hairpin and single-stranded DNA substrates. This suggests that MRE11 may expose single-stranded regions on DNA DSB. This may promote the use of HR or may activate separate pathways related to NHEJ, called microhomology mediated end joining (MMEJ) and single-strand annealing (SSA) (see Fig. 5–8).
Although DNA DSB repair defects and increased radiosensitivity have been reported for a DNA-PKcs–deficient human glioblastoma tumor cell line, there are no human syndromes attributed to defects in DNA-PK protein function. The relative levels of DNA-PKcs protein are generally lower in rodent than in human tissues, and DNA-PKcs and KU80 protein expression varies widely among different tissue types. Evidence suggests that there is no simple relationship between tumor cell radiosensitivity and the absolute level of ATM or DNA-PK protein expression (Chan et al, 1998).
Unlike HR, NHEJ does not require homology and the NHEJ proteins simply link the ends of DNA breaks together; this usually results in the loss or gain of a few nucleotides during modification of the damaged DNA to produce ligatable ends (5′-phosphate and 3′-hydroxyl). NHEJ is therefore an error-prone pathway, but is operational throughout the cell cycle. There is evidence that RAD52 (a HR-related protein), and the KU70/80 heterodimer, a DNA end-binding protein that functions in NHEJ, compete for binding to DSBs and channel the repair of DSBs into HR or NHEJ respectively, depending on the cellular context (van Gent et al, 2001). The BRCA1 and 53BP1 proteins are also suggested to direct the choice of DNA repair pathway (Bouwman et al, 2010).
5.3.6 DNA Crosslink Repair: Fanconi Anemia Proteins
The Fanconi anemia pathway is a specialized pathway for the repair of interstrand crosslinks that can occur during S-phase. The importance of this pathway is highlighted by patients with predisposition to cancer and deficiencies in 1 of 13 Fanconi anemia complementation (FANC) proteins. In this pathway, unique FANC proteins coordinate a repair mechanism including components of translesion synthesis, HR and NER (Fig. 5–9). When a replication fork approaches an interstrand crosslink, the DNA is not able to form an open configuration for the passage of the polymerase. The FANCM protein initially recognizes the lesion, which recruits an FA-core complex and creates a large E3 ubiquitin ligase that catalyses the ubiquitylation of FANCD2 and FANCI proteins that subsequently localize to the damage site and coordinate downstream functions of nucleases (eg, FAN1), DNA polymerases, NER components, and BRCA1/2-mediated HR (Kee and D’Andrea, 2010). Together these mechanisms lead to the restart of the replication fork, thereby preventing cell death or genomic rearrangements (Moldovan and D’Andrea, 2009).
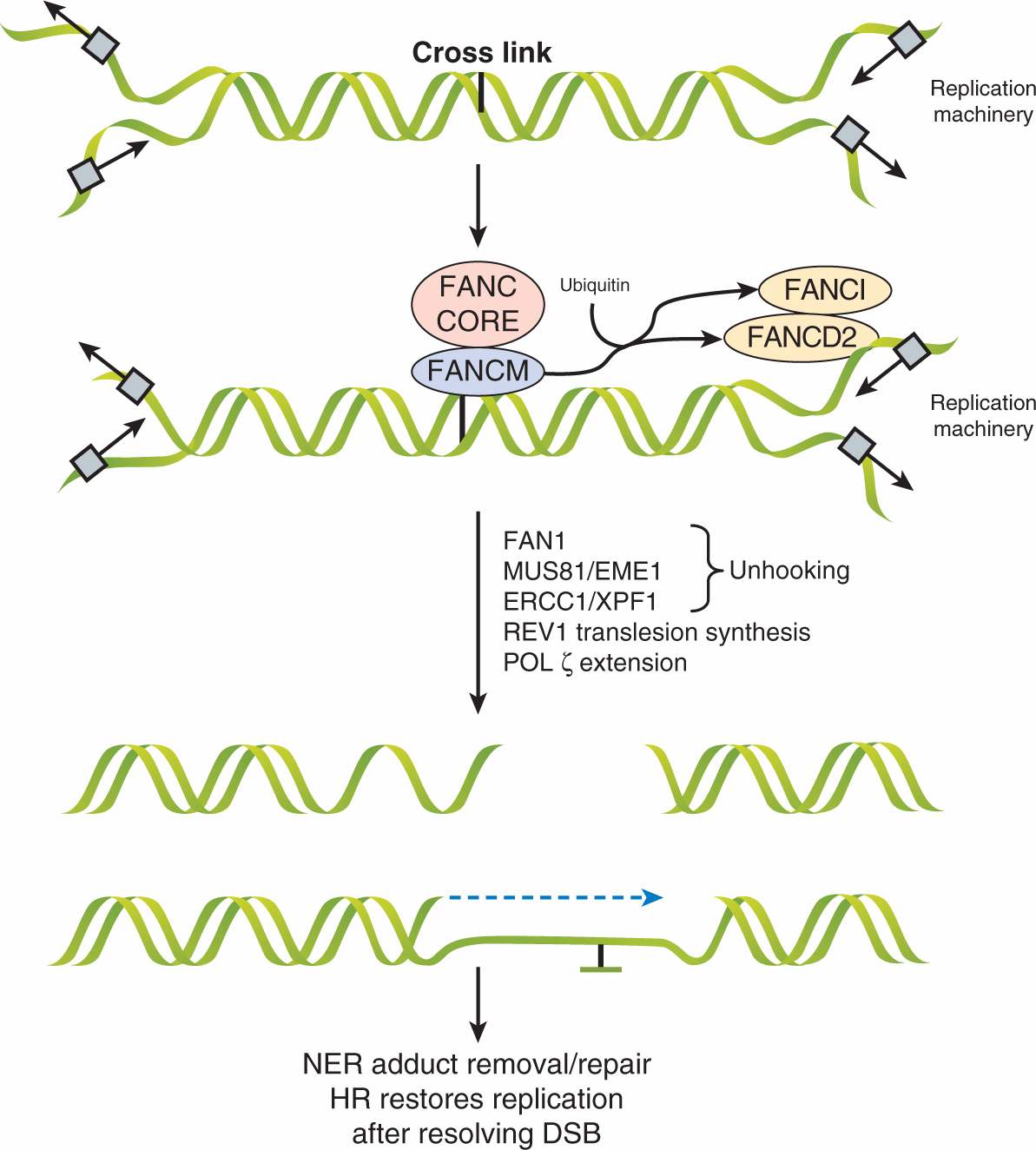
FIGURE 5–9 Fanconi anemia pathway for repairing interstrand crosslinks. Large protein complexes mediate recognition and repair of interstrand crosslinks by the Fanconi anemia pathway. Initially, when replicating DNA meets an interstrand crosslink, the FANCM protein complex recognizes the lesion and recruits the “FA core complex” consisting of FANCs A, B, C, L, E, F, and G. This induces ubiquitylation of FANCD2 and FANCI, facilitating the downstream processes. The crosslink is then unhooked by FAN1, Mus81/Eme1, and ERCC1/XPF1. Although the exact mechanism is poorly understood, Rev1 and Polz replicate across the lesion. The NER pathway removes the “flipped out” base and repairs the lesion while HR pathway components restore the replication fork after the DSB is resolved. (Adapted from Kee and D’Andrea, 2010.)
5.4 DNA DAMAGE CHECKPOINTS AND DNA REPAIR
Mammalian cells have evolved complex interrelated responses to DNA damage that, as well as direct repair, induce cell-cycle checkpoints and apoptosis. Cells can halt cell-cycle progression in the G1, S, and G2 phases at cell-cycle checkpoints to ensure that they do not enter mitosis with damaged DNA (see Chap. 9, Sec. 9.3.1). These checkpoints are initiated by kinases and are followed by a cascade of molecular events that include posttranslational modification (ie, phosphorylation, ubiquity-lation, and sumoylation) of proteins surrounding the break. There are 2 general types of cell-cycle checkpoints (Hoeij-makers, 2001; Kao et al, 2001): the mitotic spindle assembly checkpoint is responsible for ensuring that the mitotic spindle is correctly formed prior to division whereas the DNA-integrity checkpoints delay progression through the cell cycle in response to DNA damage or to defects in DNA replication (ie, G1 to S, intra-S and G2 to M; see also Chap. 9, Sec. 9.3) (Nelson and Kastan, 1994). Three related kinases, ATM, ATM-and RAD3-related (ATR) and DNA-PKcs, all sense DNA DSBs and function in DNA repair and also participate in blockade of the cell cycle (Perkins et al, 2002).
A concerted response to DNA DSBs includes the localization of a large number of proteins to the break site to concentrate kinase activity on substrates. Figure 5–10 outlines the DNA DSB-sensing pathways that lead to phosphorylation of downstream effector proteins. The ATM protein is held inactive in the cell as a dimer until DNA damage leads to autophosphorylation of ATM monomers leading to its kinase activity, both of which are enhanced by the MRN complex (Bakkenist and Kastan, 2003). This activation of ATM (and simultaneous activity of DNA-PKcs) induces phosphorylation of the histone protein H2A.X at Serine-139 forming γH2AX (see Sec. 5.6), which serves as a platform for the assembly of other components, including MDC1. ATM phosphorylation of MDC1 triggers recruitment of the RNF8 E3-ubiquitin ligase, which cooperates with UBC13 to ubiquitylate H2AX and H2A histones (Huen et al, 2007; Kolas et al, 2007; Mailand et al, 2007). In turn, the RNF168-UBC13 ubiquitin ligase complex is recruited to extend ubiquitin chains (Doil et al, 2009; Stewart et al, 2009). These events are thought to modify chromatin in such a way that proteins, including 53BP1 and the RAP80-BRCA1 complex can associate along the break site. Furthermore, downstream effectors, such as p53 and CHK2, also localize to these sites, a process that appears to be mediated by protein–protein interactions of upstream components (Al-Hakim et al, 2010; Panier and Durocher, 2009). This model supposes that the local concentration of kinase activity and downstream effector proteins contributes to the initiation of cell-cycle checkpoints, and also facilitates repair mechanisms discussed above. Many of these proteins have been found to colocalize in a spatiotemporal manner at IR-induced nuclear foci as shown in Figure 5–11 for γH2AX and 53BP1.
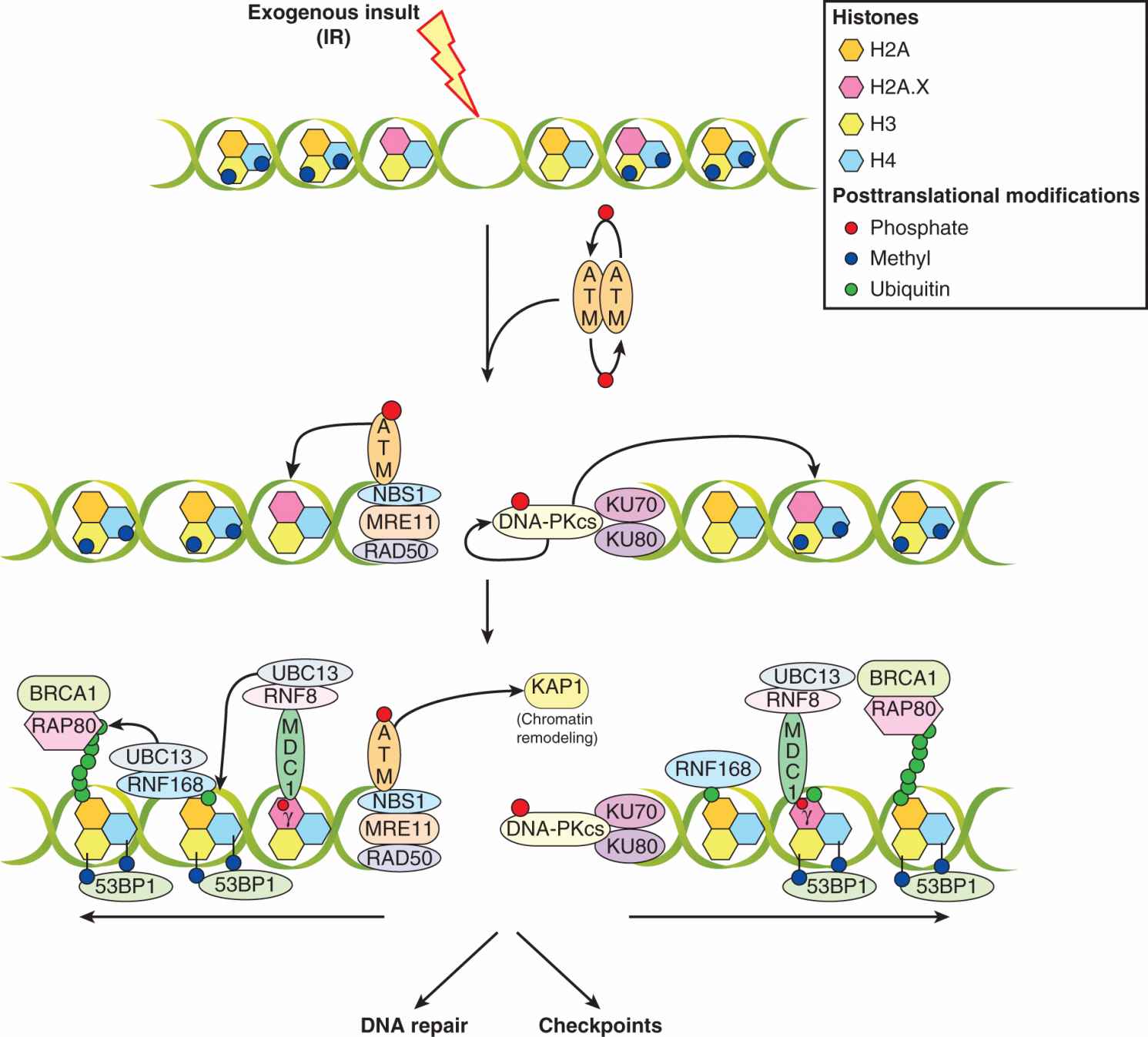
FIGURE 5–10 Signaling pathways in response to DNA DSBs induced by ionizing radiation. Signaling pathways that recognize DNA DSBs are multifaceted and likely include overlapping functions that serve to ensure checkpoints are activated even when some components do not respond properly. In addition to their role in checkpoints, these cascades also contribute to repair. When ionizing radiation (IR) creates DSBs, it leads to activation of the ATM kinase by autophosphorylation and dissociation into monomers from the inactive dimer; this process is facilitated by the MRN complex. Simultaneously, the DNA-PK complex (DNA-PKcs/KU70/KU80) (see Sec. 5.3.5) is activated, and together with ATM, phosphorylates the histone H2AX at serine-139 (forming δH2AX) in megabase domains spanning the break. This serves as a platform for the assembly of mediator of DNA damage checkpoint 1 (MDC1) which is also phosphorylated by ATM. This then recruits the E3 ubiquitin ligase RNF8, which, along with the E2-ubiquitin ligase, UBC13, ubiquitylates H2A and H2AX. This further recruits the RNF168 E3-ubiquitin ligase that, through poorly understood mechanisms, leads to the recruitment of RAP80/BRCA1 and the p53-binding protein 1 (53BP1). Multiple protein–protein interactions, facilitated by “mediator” proteins (ie, MDC1, RNF8/168, 53BP1) surrounding the DSB are thought to concentrate the “effector” proteins (ie, p53, etc) in the vicinity of “sensor” proteins (ie, ATM, MRN) to facilitate the enactment of downstream processes, including DNA repair and checkpoints.
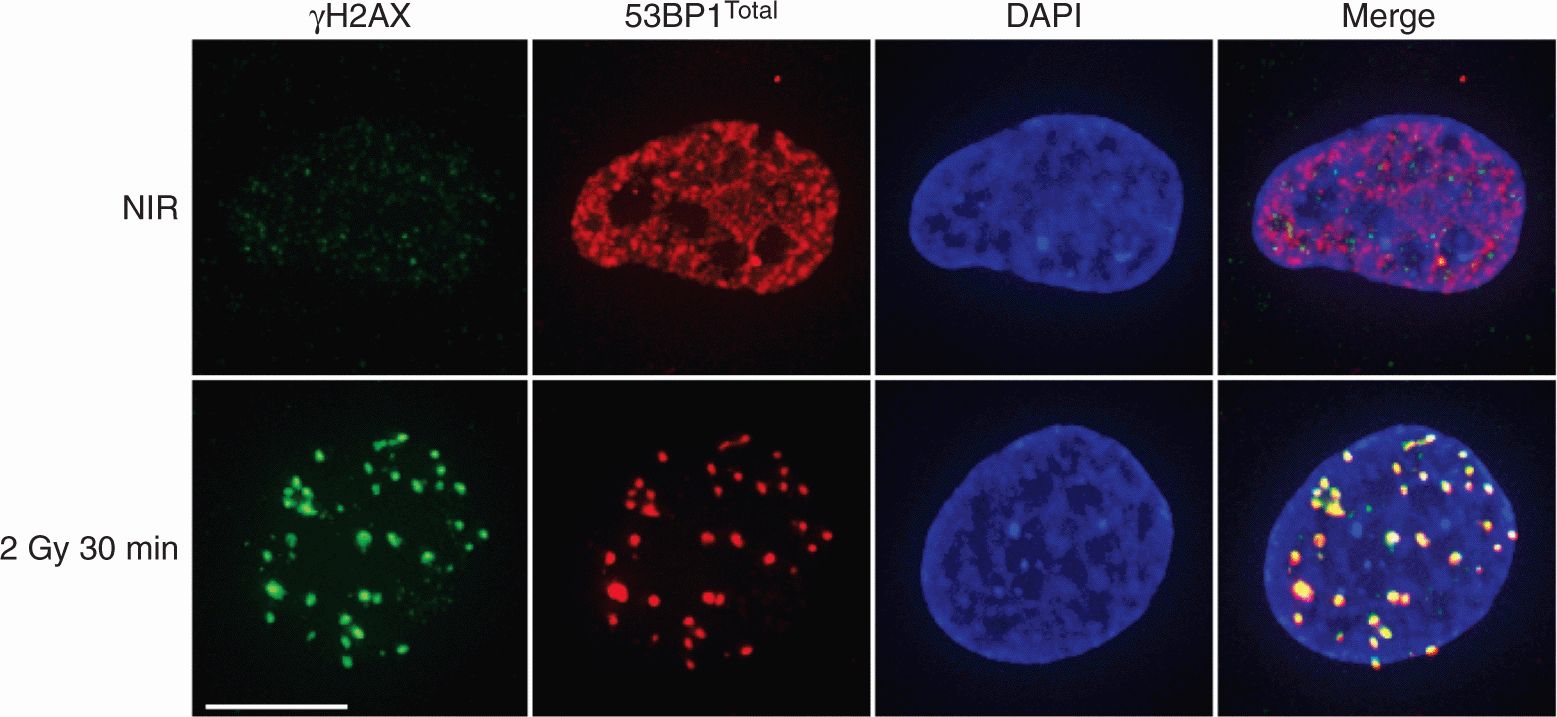
FIGURE 5–11 DNA damage-induced foci and the H2AX response. Confocal immunofluorescent images of normal human fibroblasts (GM05757) either untreated (nonirradiated [NIR]) or irradiated with 2 Gy and fixed at 30 minutes post-IR. Cells were stained for phosphorylated H2AX (δ H2AX) and total 53BP1. IR induces δ H2AX intranuclear foci whereas 53BP1 redistributes from a pan-nuclear nonnucleolar pattern into discrete foci that colocalize with δ H2AX in the DAPI-stained nucleus (shown in blue) as shown by yellow coloration. Scale bar is 10 μm. Many DSB-responsive proteins form foci in this manner (eg, MRE11, ATM, MDC1).
As described above, some lesions stall replication forks during S-phase and expose ssDNA tracts. To protect them from degradation, RPA rapidly coats this DNA and leads to recruitment of ATR interacting protein (ATRIP) and ATR. ATR and ATM are crosstalk partners and phosphorylate many of the same downstream substrates leading to cell-cycle checkpoints as described below and shown in Figure 5–12. Whether the DNA DSB response is mediated by ATM or ATR many of the same signaling cascades are activated to cause localization of downstream factors near the break (Bensimon et al, 2011). Importantly, the ATR-ATM pathways are likely not to be as distinct as once believed as recent evidence shows that ATR can activate ATM and vice-versa (Jazayeri et al, 2006; Stiff et al, 2006). Recent phosphoproteome experiments have shown that they share upwards of 700 protein targets, indicating that these pathways are heavily intertwined (Matsuoka et al, 2007). These overlapping pathways increase the likelihood that sufficient checkpoint responses will take place.
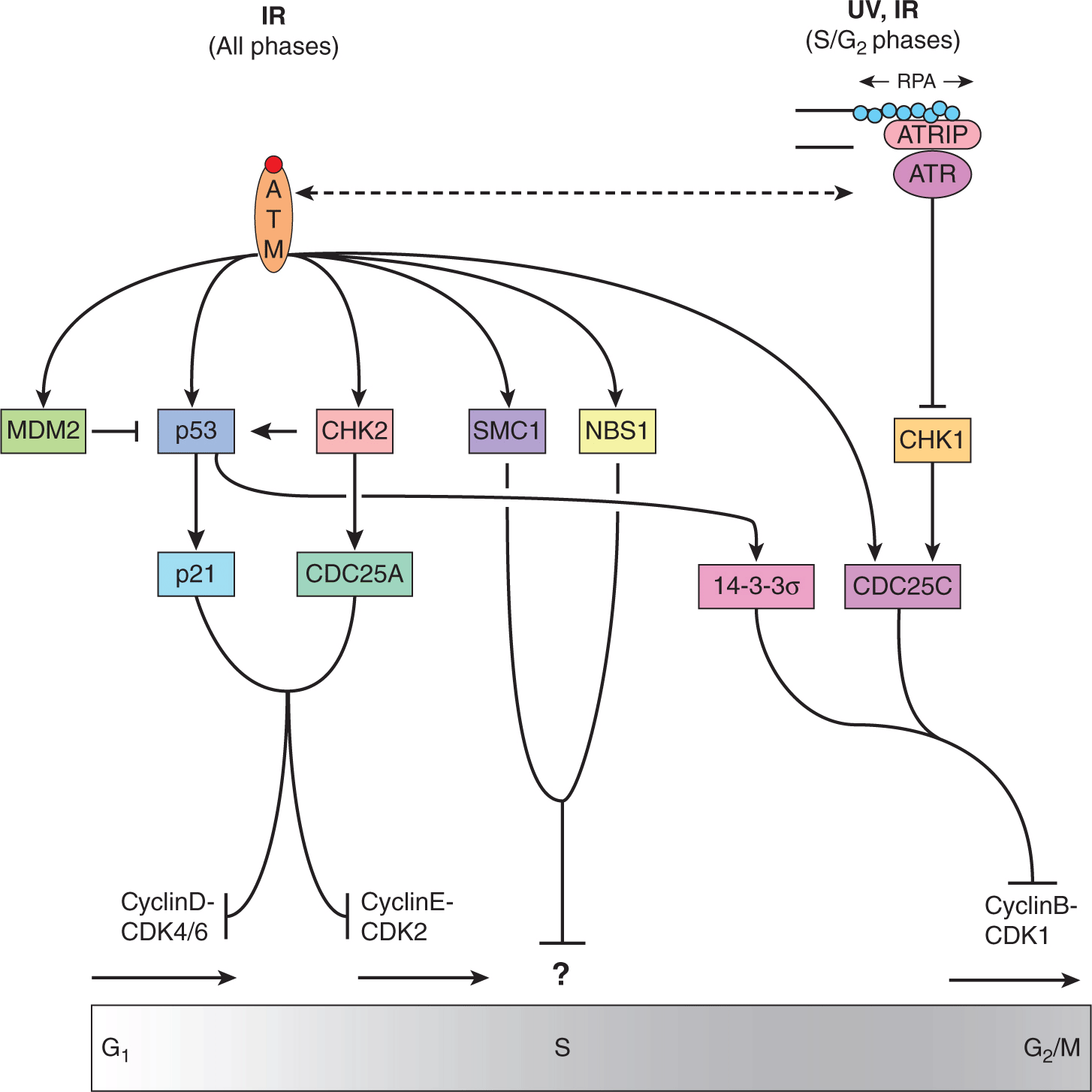
FIGURE 5–12 Cell-cycle checkpoint activation downstream of ATM and ATR. Induction of DSBs by IR activates ATM, which phosphorylates MDM2 preventing p53 proteasomal degradation. ATM also activates, via phosphorylation, CHK2, which, in turn, phosphorylates CDC25A to inactivate its phosphatase activity and block CyclinE-CDK2-Rb– mediated S-phase entry. The stabilization of p53 is accompanied by ATM- and CHK2-phosphorylation which activate transcription of p53 target genes, such as p21. As a CDK inhibitor p21 blocks CyclinD-CDK4/5 and CyclinE-CDK2 and, therefore, S-phase entry. Activated ATM also phosphorylates SMC1 and NBS1 proteins that slow progression through S-phase by poorly defined mechanisms. DNA damage incurred during S-phase (by IR or UV) may create ssDNA. This ssDNA is coated by RPA recruiting ATR via its coactivator, ATRIP. Active ATR phosphorylates CHK1, which inactivates CDC25C. ATM activity and p53-induction of 14-3-3σ also inactivate CDC25C. Inactive CDC25C cannot remove inhibitory phosphate groups on CDK1 and therefore blocks CyclinB-mediated transition to mitosis. Crosstalk between ATM and ATR leads to simultaneous activation of many of these pathways, as described in the text.
The colocalization of ATM activity with effectors initiates a G1-to-S cell-cycle checkpoint by a posttranslational stabilization of the p53 protein via direct phosphorylation of serine 15 on p53 (Canman et al, 1998). ATM also phosphorylates threonine residue 68 on the Chk2 protein, which can, in turn, phosphorylate p53. These phosphorylations lead to p53 nuclear accumulation by interfering with a nuclear export site contained within the amino terminus of the p53 protein and by preventing degradation of p53 by MDM2 (see Chap. 7, Sec. 7.6.1; Liang and Clarke, 2001). Because p53 acts primarily as a transcription factor, current models regard its stabilization following DNA damage as a mechanism to activate the cyclin D/E-kinase complex inhibitor, p21WAF, which leads to continual hypophosphorylation of the Rb protein to effect G1 cell-cycle arrest. A second G1 checkpoint pathway is also activated that targets the CDC25A phosphatase that is essential for G1/S transition (Falck et al, 2001).
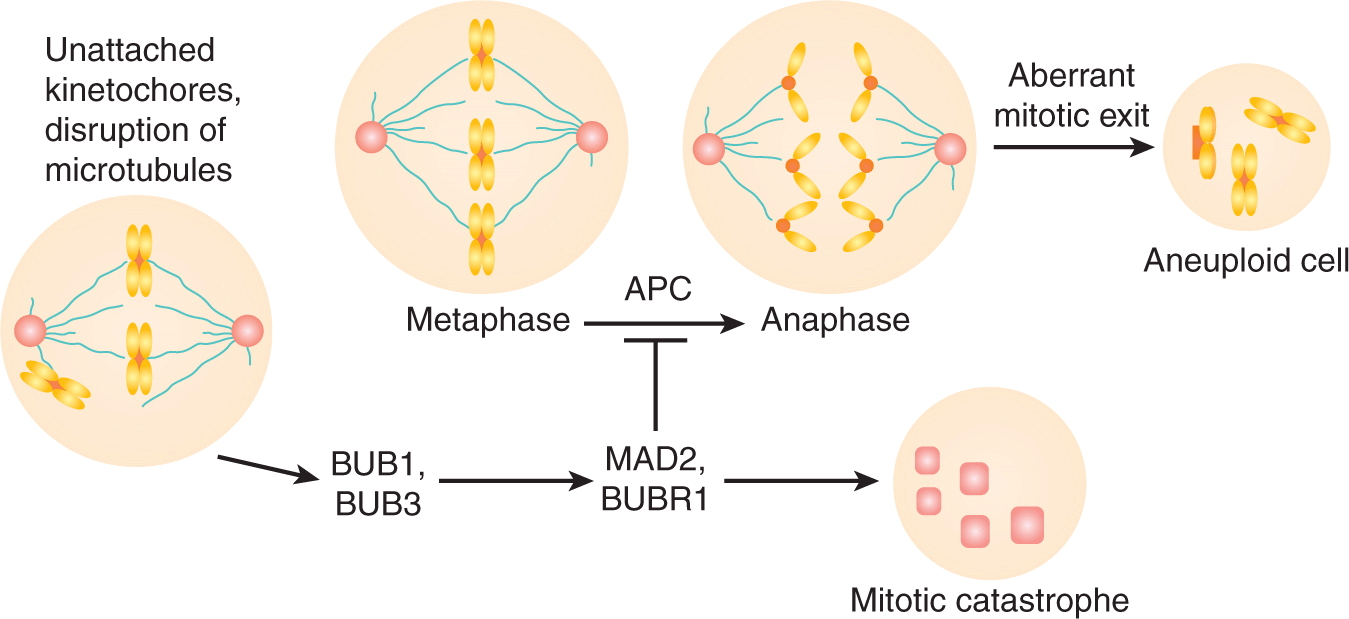
FIGURE 5–13 Improper chromosome alignment on the mitotic spindle can activate the spindle checkpoint mediated by the BUB1, BUB3, BUBR1, and MAD2 proteins that localize to kinetochores. An intact spindle checkpoint induces metaphase arrest through inhibition of the APC. Defective spindle-checkpoint function results from either loss of BUB1- and BUB3-dependent signaling or abrogation of MAD2, BUBR1-mediated inhibition of the APC. This leads to the absence of a functional mitotic spindle checkpoint generating aneuploid cells. (Adapted from Stewart et al, 2003.)
Further important targets of ATM-mediated phosphorylation are the BRCA1, NBS1, and FANCD2 proteins, which initiate both DNA repair and an S-phase DNA damage checkpoint (Cortez et al, 1999; Gatei et al, 2000; Lim et al, 2000). The ATM-mediated phosphorylation of NBS1 seems to be required for proper execution of an S-phase checkpoint as mutations in the ATM-associated serine residues (ie, Ser278, -343, or -397) of NBS1 leads to radioresistant DNA synthesis, a phenotype associated with AT cells.
Following their activation by ATM and ATR, CHK1 and CHK2 phosphorylate a conserved site (Ser216) on protein phosphatase CDC25C, which results in it being inactivated and bound by the 14-3-3σ protein. The inactive CDC25C is then incapable of removing an inhibitory phosphate group on Tyr-15 of CDC2, preventing entry into mitosis. In addition, p53-dependent transcriptional repression of the CDC2 and cyclin B promoters may contribute to the maintenance of the G2/M checkpoint in mammalian cells, which may allow for repair of chromosomal or chromatid damage prior to cellular division. The elegant crosstalk between ATM and ATR and factors involved in DNA repair, DNA damage signaling and cell-cycle control act to maintain genomic stability and inhibit carcinogenesis (see Fig. 5–12).
Chromosome segregation is crucial for cells to maintain the integrity of their genome (Jasin, 2000). Mitotic exit occurs after ubiquitination and proteolytic degradation of cyclin B by the anaphase promoting complex (APC), which inactivates CDK1. The mitotic spindle checkpoint monitors the interaction between chromosomes and microtubules at highly specialized chromosomal regions called kinetochores (Musacchio and Hardwick, 2002). This checkpoint delays chromosome segregation during anaphase to correct any defects in the mitotic spindle apparatus; if defects persist, the cell undergoes cell death. The kinetochore-associated MAD2, BUBR1, BUB1, and BUB3 proteins are critical constituents of the spindle-checkpoint pathway: MAD2 and BUBR1 regulate mitotic progression by direct interaction and inhibition of the APC machinery and BUB1 and BUB3 also mediate mitotic arrest after disruption of microtubules as shown in Figure 5–13. Cells that lack either BUB1 or BUB3 do not undergo mitotic arrest when treated with spindle-disrupting agents, such as the chemotherapy drugs docetaxel or vinblastine.
Genetic defects in the spindle checkpoint can lead to chromosome loss during mitosis and meiosis with links to the pathogenesis of several human tumors. For example, in human colon and breast carcinoma cells, BUB1 mutations have been identified that facilitate the transformation of cells that lack BRCA2. In other studies, MAD2 haploinsufficiency significantly elevated the rate of lung tumor development in MAD2+/– mice compared with age-matched controls (Stewart et al, 2003). There is potential for therapeutic intervention in cancer treatment by abrogation of cell cycle checkpoints using agents targeted at particular kinases (ie, UCN-01 for CHK1) in cells that already lack other checkpoints (ie, p53 mediated). This results in catastrophic mitotic death as a result of cell progressing into mitosis with unrepaired DNA DSBs (Lapenna and Giordano, 2009).
5.5 HUMAN DNA REPAIR DISORDERS AND FAMILIAL CANCERS
As discussed above, repair of DNA plays an important role in determining cancer predisposition. Functional analyses of DNA repair proteins and their pathways in vivo have benefited from molecular recombinant technologies whereby a specific DNA repair gene is either aberrantly expressed or rendered null (ie, gene knockout) in murine embryonic stem cells (see Chap. 2, Sec. 2.4.5). Some, but not all, DNA repair proteins are required for mammalian development as a number of the mice were embryonic lethal (ie, mice null for Rad51 and Rad50). Furthermore, repair-deficient mice are also prone to chromosomal instability and to the early onset of specific cancers (Hakem, 2008).
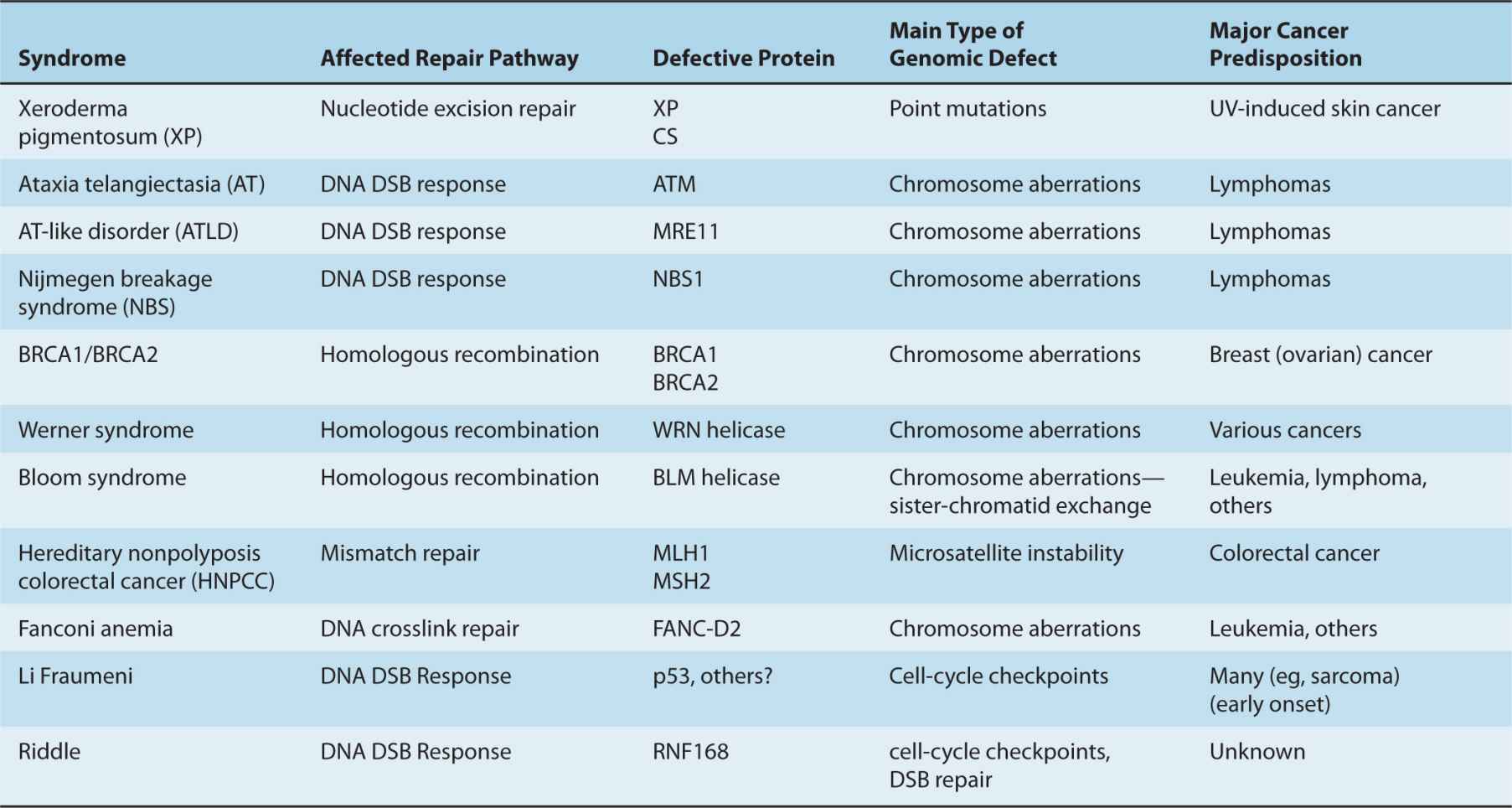
Several human disease syndromes are associated with pronounced cellular sensitivity to DNA-damaging agents because of hereditary deficiencies in DNA repair or in signaling pathways that are activated by DNA damage (Table 5–1). People with several of these syndromes show marked chromosomal instability and predisposition to malignancy, highlighting the importance of genetic maintenance for prevention of carcino-genesis. Some of these disorders are discussed below.
TABLE 5–1 Cancer prone human syndromes with defective DNA repair.
5.5.1 Ataxia Telangiectasia The AT syndrome is an autosomal recessive disease characterized by cerebellar degeneration, immunodeficiency, chromosomal instability, cancer predisposition, radiation sensitivity (see Chap. 15, Sec. 15.4.1 and Figure 15–16), and cell-cycle abnormalities (Rotman and Shiloh, 1998; Weissberg et al, 1998). A mutation in a single gene coding for ATM is responsible for this syndrome. The ATM gene encodes a large protein that possesses a highly conserved C-terminal kinase domain related to PI3K (see Durocher and Jackson, 2001). Most mutations in ATM result in truncation and destabilization of the protein, but certain missense and splicing errors produce a less-severe phenotype. The pleiotropic clinical symptoms associated with AT may be explained partly by modification of cellular metabolism. For example, the loss of ATM function is associated with increased oxidative damage, particularly in cerebellar Purkinje cells, implicating a role for ATM in the response to reactive oxygen species and possibly explaining the cerebellar degeneration observed in AT-affected individuals. These observations support the hypothesis that ATM is associated with oxidative metabolism and the possible prevention of the DNA-damaging effects of reactive oxygen species (Rotman and Shiloh, 1998).
The association between mutation of the ATM gene and a high incidence of lymphoid malignancy in those patients with AT, together with the development of lymphoma in ATM-deficient mice, supports the hypothesis that inactivation of the ATM gene is important in the pathogenesis of sporadic lymphoid malignancy. LOH at 11q22-23 (the location of the ATM gene) is a common event in lymphoid malignancy. Frequent inactivating mutations of the ATM gene have been reported in patients with Hodgkin and non-Hodgkin lymphoma, rare sporadic T-cell prolymphocytic leukemia (T-PLL), B-cell chronic lymphocytic leukemia (B-CLL), and, most recently, mantle cell lymphoma (MCL). Furthermore, AT heterozygotes may have a slightly increased risk of breast cancer when compared to the normal population. These data suggest that ATM functions as a tumor-suppressor gene (Rotman and Shiloh, 1998).
Cells derived from AT patients and ATM–/– mice in culture display hypersensitivity to ionizing but not UV radiation; they exhibit chromosomal instability and a mild DNA DSB rejoining defect. ATM-deficient cells also have radioresistant DNA synthesis and further defects in the G1/S–, S– and G2/M–phase DNA damage checkpoints. One model suggests that ATM or ATR kinase activity requires colocalization with its substrate (ie, p53 or other proteins) at sites of DNA damage to initiate signaling within DNA repair and cell-cycle checkpoint pathways (Al Rashid et al, 2005, 2011).
A number of other diseases, including ATLD and NBS, show similar disease phenotypes to AT supporting the cooperation of their cognate molecular defects (MRE11 and NBS1, respectively) with ATM in suppression of genetic instability as discussed (see Sec. 5.4). The exact mechanism for the developmental defects in these patients is not well understood, but these syndromes have been instrumental in the understanding of the molecular pathways that control the responses to DNA damage, particularly DNA DSBs.
5.5.2 Xeroderma Pigmentosum and Related Disorders The human XP, CS, and trichothiodystrophy (TTD) disorders exhibit cellular UV sensitivity as a consequence of deficiency in NER (see Sec. 5.3.3; Hoeijmakers, 2001). For reasons that are not understood, only XP patients are cancer-prone with a dramatic 1000-fold increase in the incidence of UV-induced skin cancer. This is caused by mutations in 1 of 7 XP genes (XPA-XPG) in their cells. CS is characterized by a TCR defect secondary to mutations in CSB and CSA genes. CS patients exhibit neurodegeneration and premature aging related to inappropriate apoptosis. TTD patients share many features of CS patients, but also have brittle hair, nails, and scaly skin, secondary to reduced expression of epidermal matrix proteins.
5.5.3 Hereditary Nonpolyposis Colon Cancer Hereditary nonpolyposis colon cancer (HNPCC) is the most common form of hereditary colon cancer, accounting for 5% to 8% of all colon cancers. People with HNPCC also have an excess of endometrial, small bowel, and renal cancer. This familial cancer syndrome occurs secondary to genetic instability acquired through deficient MMR by virtue of mutations in 6 different MMR genes (see Sec. 5.3.1) (Harfe and Jinks-Robertson, 2000). Replication of repetitive DNA sequences, termed microsatellites, can result in gains or losses of these repeated units giving rise to microsatellite instability (MSI). The MMR system suppresses MSI. Cells from HNPCC patients (and also from sporadic colorectal tumors) can acquire runs of approximately 4 to 40 repeated mononucleotides or dinucleo-tides, such as TTTT or CACACA, at multiple sites within the genome as a consequence of MSI.
More than 300 different predisposing MMR genetic mutations have been documented in human cancers, mainly affecting MLH1 (approximately 50%), MSH2 (approximately 40%), and MSH6 (approximately 10%) (Friedberg, 2001). Genetically predisposed individuals with HNPCC carry a defective copy of an MMR gene in every cell and mutation rates in tumor cells with MMR deficiency are 100- to 1000-fold higher than in normal cells. Somatic inactivation of the remaining wildtype copy in a target tissue, typically colon, gives rise to a profound repair defect and increased rates of mutation in cells (ie, a “mutator” phenotype) with progressive accumulation of mutations in APC, p53, or other genes that contribute to colon cancer development (Jiricny and Nystrom-Lahti, 2000; Loeb, 1991; Peltomaki, 2001).
5.5.4 Li-Fraumeni Syndrome One of the most frequently mutated genes in human tumors occurs in that encoding the p53 protein, which is involved at multiple points in the response to DNA damage and genetic instability, including but not limited to cell-cycle checkpoints, apoptosis, and modulating DNA repair (see Chap. 7, Sec 7.6.1). The Li-Fraumeni syndrome occurs in people with germline mutations of TP53. Originally characterized in 1969 in early onset cancers in relatives of children with rhabdomyosarcoma, p53 has since been found to be mutated in more diverse tumor types and in various families with inherited cancers (Palmero et al, 2010). In people with the Li-Fraumeni syndrome, 50% develop cancer before age 40 years, and 90% develop cancer before age 60 years. These are tumors of various histopathologies and include sarcomas and brain and breast cancers. Although mutation of other genes (eg, CHK2) have been suggested to be alternative predisposing factors for Li-Fraumeni syndrome, the evidence for such involvement is controversial (Palmero et al, 2010).
5.5.5 Fanconi Anemia Fanconi anemia (FA) is a very rare autosomal recessive disorder with 1 in 300 people carrying mutations in 1 FANC gene; 1 to 5 in 1 million people are affected by this disorder (D’Andrea, 2010). Although rare, this syndrome has led to the identification of 13 “complementation group” proteins that are mutated or lost in FA patients and that comprise a distinct pathway in mammalian cells (see Sec. 5.3.6). The definitive test for FA is to treat lymphocytes from patients with diepoxybutane, a DNA crosslinking agent, and assess chromosome breakage. In FA patients, this leads to accumulation of breaks owing to an inability to repair interstrand crosslinks in DNA. Tumors (eg, leukemia, head and neck) develop in at least 20% of FA patients and this number would likely be much higher if bone marrow failure and other manifestations of the syndrome did not lead to early death of afflicted individuals.
5.5.6 BRCA1/2 Germline mutations in a single copy of the BRCA1 or BRCA2 tumor-suppressor gene occur in approximately 1 in 250 women and lead to early onset breast and ovarian cancer (Narod and Foulkes, 2004). The large majority of tumors from carriers display LOH such that both copies of the gene become mutated, rendering the cell BRCA1 or BRCA2 null. Mechanistic roles for BRCA1/2 have been observed in various DNA repair pathways including the FA pathway (see Sec. 5.3.6) and, HR (see Sec. 5.3.4), and in cell-cycle checkpoints and maintenance of ploidy (O’Donovan and Livingston, 2010). The defect in HR in BRCA1/2 tumors is being tested for a possible role in synthetic lethality using PARP inhibitors in ongoing clinical trials (Audeh et al, 2010; Tutt et al, 2010).
5.6 ASSAYS OF DNA DAMAGE AND REPAIR
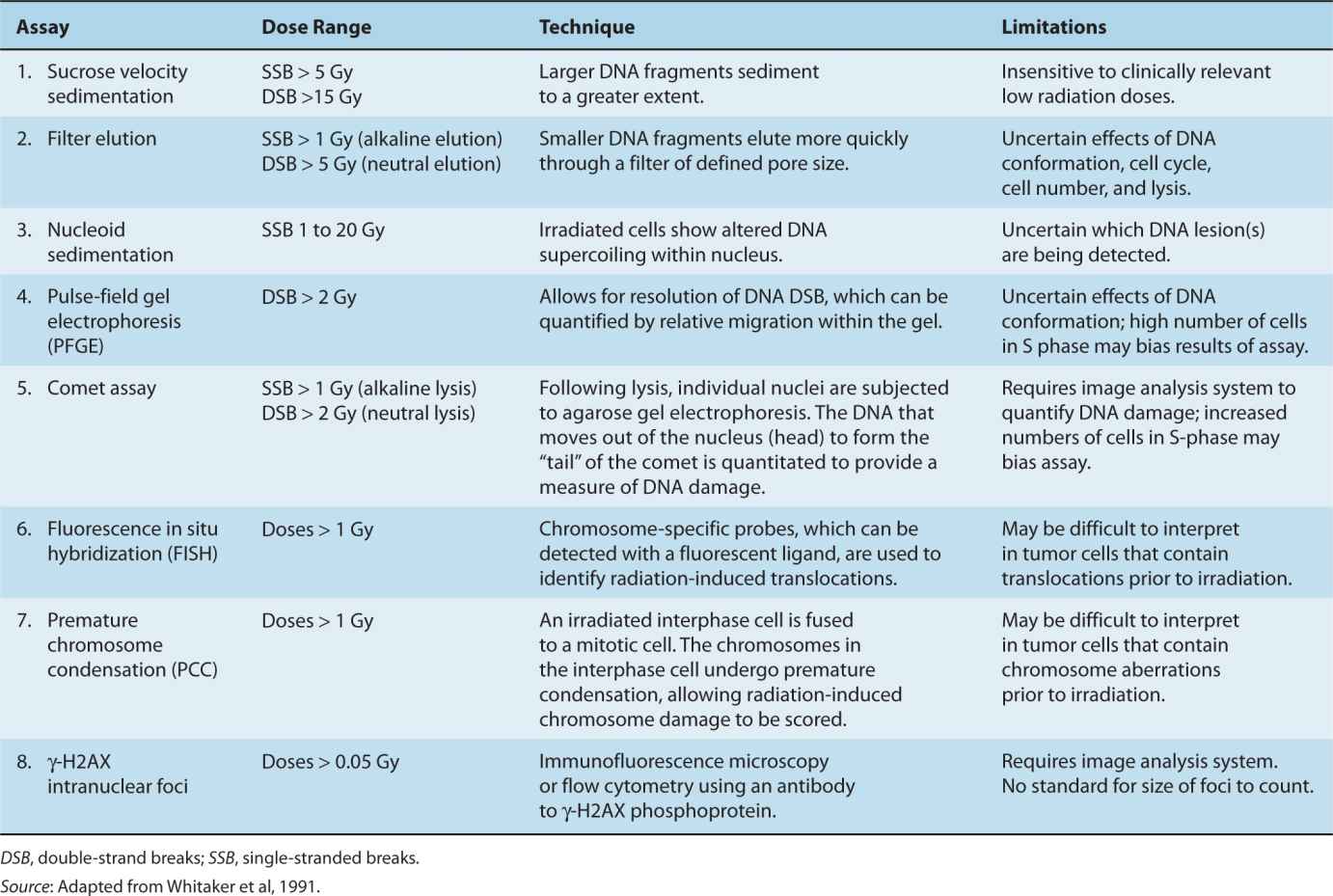
It is important to be able to measure the DNA damage that is present in a cell, and to determine how well a cell is able to repair this damage by one of the mechanisms previously described. Table 5–2 outlines techniques that have been used to measure different types of DNA strand breaks. Modifications of these assays provide the ability to measure base damage and photoproducts that result when DNA repair pathways are defective. Techniques such as velocity sedimentation, filter elution, assays for chromosomal damage, and DNA electrophoresis have been used to study specific DNA lesions caused by radiation (Fairbairn et al, 1995; Whitaker et al, 1991). Two techniques, fluorescence in situ hybridization (FISH; see Chap. 2) and premature chromosome condensation (PCC), allow the quantification of single- or double-strand breaks following doses of IR as low as 1 Gy (Sasai et al, 1994). Other techniques, such as pulsed-field gel electrophoresis (PFGE) or the Comet assay (Fairbairn et al, 1995), can facilitate the separation and quantification of large DNA fragments secondary to single- or double-strand DNA breaks following radiation, but, with exception of the comet assay, require much higher (>10 to 20 Gy) doses because of their lower sensitivity. DNA DSBs are of particular importance as they are the most lethal form of DNA damage (see Chap. 15).
TABLE 5–2 Assays for the detection of DNA damage following ionizing radiation.
IR leads to rapid phosphorylation of nucleosomal histone protein, H2AX. γH2AX is the phosphorylated form that can be quantified using a specific antibody as an intracellular marker of DNA DSBs (Rogakou et al, 1999; see Figs. 5–10 and 5–11). This early event precedes the actions of repair enzymes involved in HR and NHEJ of these breaks. Microscopically visible nuclear foci, each containing thousands of γH2AX molecules covering about 2 Mb of DNA surrounding the break, can be detected using antibody staining and fluorescence microscopy. The number of γH2AX foci has been directly correlated to the number of DNA DSBs in 125IUdR-treated cells as each 125I decay yields a DNA DSB and each DNA DSB yielded a visible γH2AX focus (Sedelnikova et al, 2002). Following IR these foci resolve with kinetics that correlate with other biochemical DSB rejoining assays (eg, PFGE) such that approximately 50% of breaks remain after 2 hours and by 24 hours following the radiation dose almost all breaks are rejoined. It is probable that increased residual nuclear foci (≥24 hours post-IR) represent non-rejoined DNA DSBs and lead to subsequent cell lethality (MacPhail et al, 2003). However, there are confounding factors that activate γH2AX, such as apoptosis and cell-cycle and oncogene signaling (Lobrich et al, 2010). This method has also been adapted to detect other types of lesions, such as 6-4PPs, to which antibodies have been generated (Vermeulen, 2011).
Using innovative microscopic and fluorescent technologies, researchers can specifically generate and visualize discrete DNA DSBs. For example, use of high fluence targeted laser microirradiation induces DNA DSBs in a spatially restricted manner within the nucleus. These can be used to monitor the recruitment of repair proteins to DNA damage, and can also be used in combination with more advanced techniques such as fluorescence recovery after photobleaching (FRAP) to determine the residence of repair proteins at the DNA damage site in live cells. Despite the utility of such methods for localization of DNA damage, there remains to be determined the exact nature of the damage created by these techniques, and conclusions drawn from such assays are typically bolstered by the more traditional techniques outlined in Table 5–2.
5.7 REGULATION OF TELOMERE LENGTH AND CANCER
Human cells have evolved a complex network of proteins that bind to chromosomal ends, called telomeres, to protect them from being inappropriately recognized as DNA damage. Disruption of this nucleoprotein complex, either through loss of telomere DNA or direct disruption of protein function at the telomere, signals a DNA damage response leading to cell death or cessation of cell division. Cells that retain the capacity to proliferate, such as stem cells and cancer cells, must thus find a mechanism to maintain telomere integrity. Most often, cancer cells achieve this protection through the activation of an enzyme that adds new telomere DNA onto chromosome ends, called telomerase. More rarely, maintenance of telomeres is achieved through recombination, exchange, or copying of existing telomere DNA tracts through mechanisms collectively referred to as “alternate lengthening of telomeres” (ALT). Together, telomerase and the nucleoprotein complex that protects telomeres ensure the maintenance of telomere integrity in dividing mammalian cells.
Pioneering genetic experiments in the fruit fly Drosophila melanogaster and in maize laid the foundation for our appreciation of the importance of telomeres in genome stability (reviewed in Greider, 1996). These initial experiments determined that DNA damage could result in the loss of the terminal “knob” of linear chromosomes. Loss of telomeres in maize led to fusion or loss of chromosomes and an ensuing cycle of chromosome breakage, chromosome fusion, and anaphase bridges during cell division that is referred to as the breakage-fusion-bridge cycle. We now appreciate that telomere integrity is critical to the viability of normal and malignant cells in many organisms, and that loss of telomere DNA is an important contributor to genomic instability.
5.7.1 Telomeres Protect Chromosomal Ends from Recognition as DNA Damage
Most linear chromosomes terminate in a long, noncoding repetitive tract of G-rich, telomeric-DNA that varies in its sequence and average length between organisms. In humans, telomeres are composed of 4 to 15 kilobase pairs (kbp) of the hexanucleotide sequence 5′-TTAGGG-3′ followed by 100 to 150 nucleotides of a single-stranded TTAGGG-3′ overhang. The 3′ single-stranded overhang is looped back on itself in a structure termed the t-loop (reviewed in de Lange, 2004). Internal to the telomeric tract there are several kilobases of degenerate telomeric sequence, called subtelomeric DNA. The subtelomeric and telomeric regions exist in a nucleoprotein complex containing several telomere-binding proteins and chromatin-associated proteins. This nucleoprotein complex possesses a distinct nucleosomal structure that is thought to protect the chromosome ends from being perceived as damaged DNA, although in response to uncapping of telomere DNA overt chromatin remodeling is not observed (Wu and de Lange, 2008; Fig. 5–14). Telomere dysfunction, either by erosion of telomere DNA or through loss of telomere end protection, can trigger the recruitment of several DSB response factors, including γH2AX, 53BP1, MRE11/RAD50/NBS1, and phosphorylated ATM. These resultant telomere-dysfunction–induced foci (TIF) coincide with the activation of the checkpoint kinases ATR and/or ATM, which, in turn, phosphorylate Chk1 and Chk2 and lead to p53- and p21-dependent cell apoptosis and cycle arrest (reviewed in d’Adda di Fagagna, 2008). Thus, many factors that play a role in the DNA damage response elsewhere in the genome also play important roles at the telomere.
To prevent the gradual erosion of linear DNA that arises as a result of the inability of DNA polymerases to completely replicate the 5′ ends of linear DNA, a special cellular mechanism must compensate for telomere loss. This mechanism entails the addition of new G-rich, single-stranded telomere DNA to chromosome ends by telomerase, which carries its own telomere-complementary RNA template (see Fig. 5–14). This enzyme employs a reverse transcriptase (telomerase reverse transcriptase [TERT]) and the RNA template (telomerase RNA-TR) to direct new telomere DNA synthesis, 1 nucleotide at a time, onto chromosome ends (reviewed in Greider and Blackburn, 1996).
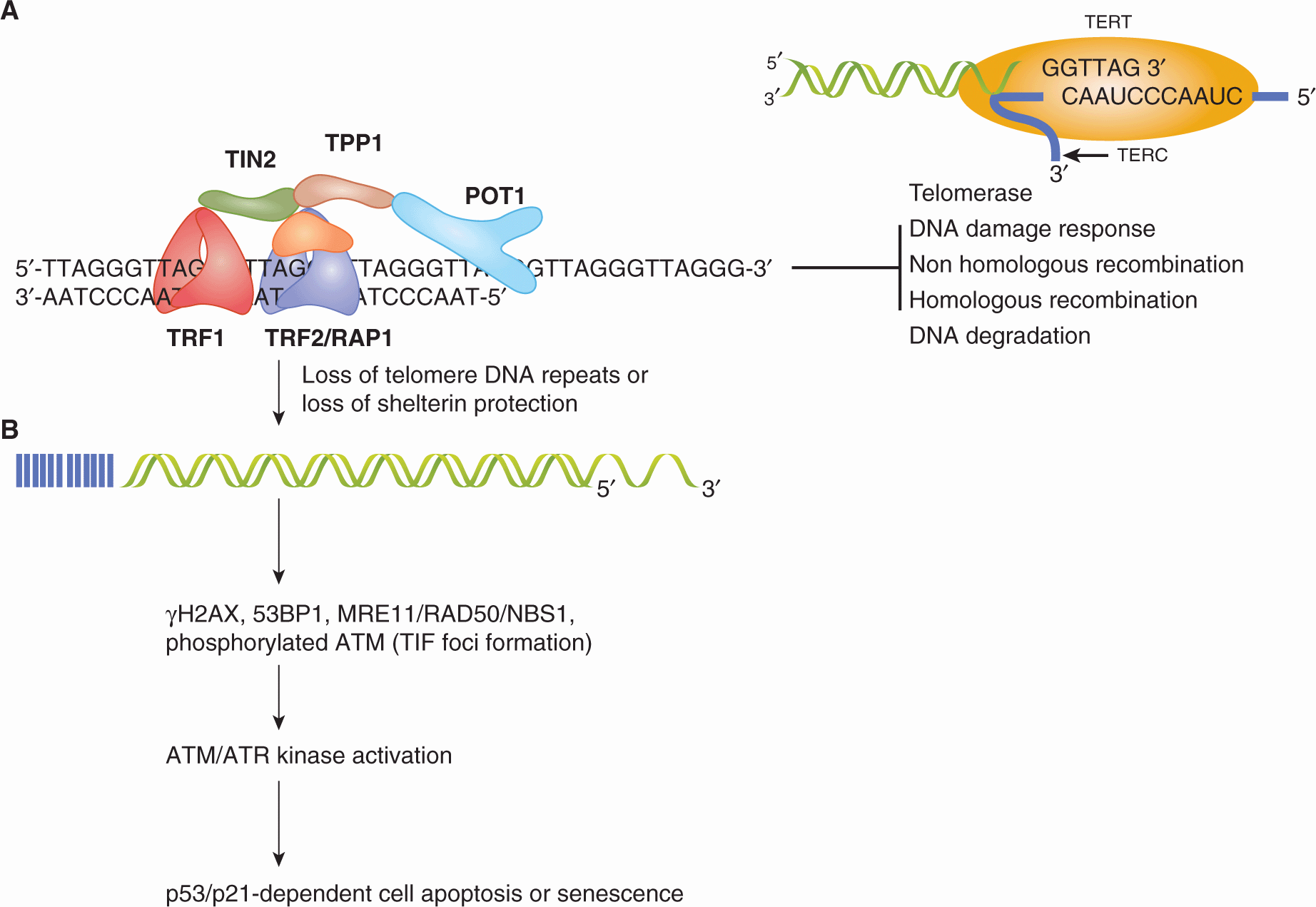
FIGURE 5–14 Telomere maintenance by telomerase and shelterin and the consequences of telomere dysfunction. A) Telomere DNA, telomerase, and shelterin (TIN2, TRF1, TRF2, POT1, RAP1, TPP1). Telomeres protect against DNA damage and degradation. The access of telomerase is limited by telomere-bound POT1 and TRF1. B) Dysfunctional telomeres that arise via loss of telomere DNA repeats or loss of protection of shelterin induce DNA damage foci formation in telomeres (TIF) and activate ATM or ATR kinases leading to p53/p21-dependent cell apoptosis, cell-cycle arrest, and cellular senescence. (Reproduced with permission from Lue NF, Autexier C. Telomerases: Chemistry, Biology and Clinical Applications. John Wiley and Sons; 2012.)
Telomeric sequences added by telomerase compensate for the gradual erosion of chromosome ends to enable cells undergoing multiple divisions during cell proliferation such as stem cells and cancer cells to maintain telomere length and chromosome stability. New telomere DNA synthesis is coordinated with replication of the telomeric DNA tract by conventional DNA polymerases, and, indeed, telomerase interacts with subunits of the DNA replication machinery (reviewed in Price et al, 2010). Concomitant with or following replication, the 5′ and 3′ ends of telomeric DNA are processed via enzymatic pathways that have not yet been fully elaborated. During de novo telomere DNA synthesis by TERT, the telomerase RNA template is reversed transcribed into telomeric DNA (see Fig. 5–14). The assembly of active telomerase in vivo requires other components. For example, dyskerin and its protein partners NHP2, NOP10, and GAR1 bind to the 3′ terminal H/ACA box on telomerase RNA to modulate telomerase assembly and stability (Egan and Collins, 2010). TCAB1 (telomerase Cajal body protein 1) interacts with dyskerin and recognizes the CAB box in the telomerase RNA, also facilitating telomerase assembly and trafficking to Cajal bodies (reviewed in Venteicher and Artandi, 2009). An excellent inventory of telomerase subunits from various organisms can be found at: http://telomerase.asu.edu/.
5.7.2 Telomere Regulation in Normal Cell Proliferation
In humans, normal cells often express low or undetectable levels of telomerase activity because of transcriptional repression of the TERT catalytic protein subunit of telomerase, and therefore undergo telomere shortening with each subsequent cell division (reviewed in Greider, 1998b). Normal cells do not replicate indefinitely in culture. The eventual loss of replica-tive potential is termed cellular senescence or the Hayflick limit. The time to the cessation of cell division correlates closely with a critically eroded telomere at which telomeric tracts can no longer be detected (called a telomere signal-free end [TSFE]; reviewed in Greider, 1998b). In the absence of the tumor-suppressor genes, p53 or pRb, cells can undergo additional population doublings before eventually reaching a proliferative block, called crisis. At crisis, cells exhibit extensive genome instability and most will undergo apoptosis. Only a small fraction of cells (approximately 1 in 107) survive by either activating expression of the endogenous hTERT gene, or via other telomerase-independent mechanisms, such as ALT. Stem cells from various human tissues express telomerase during proliferative bursts, although usually at levels sufficient only to maintain telomere integrity and insufficient to elicit overall telomere lengthening (reviewed in Blasco, 2007).
The hypothesis that cellular senescence is causally linked to “critically short” telomeres is supported by a tight correlation between them, and the finding that reintroduction of TERT into human primary cells renders them capable of indefinite proliferation; indeed reintroduction of this single gene indefinitely prolongs the life span of human cells in culture (Bodnar et al, 1998; Vaziri and Benchimol, 1998). Despite being “immortal” in a culture dish, TERT-reconstituted human cells appear to preserve their normal morphology, response to external stress, and karyotype (Vaziri et al, 1999). Thus, TERT itself does not possess the classical characteristics that define an oncogene (Greider, 1998a), although in combination with other oncogenes TERT promotes the malignant transformation of normal cells (reviewed in Hahn, 2002).
In mice, disruption of the genes encoding the telomerase RNA or TERT leads to telomere shortening in all tissues (reviewed in Liu and Harrington, 2012). For example, following several generations of breeding in the absence of the telomerase RNA, visible defects are observed in several organs and stem cell compartments in the germline, blood, and gut (Lee et al, 1998). Critically short telomeres lead to telomeretelomere fusions, which in turn activate a p53-dependent apoptotic response (Chin et al, 1999; Lee et al, 1998). Crossing telomerase-deficient mice with p53-deficient mice can extend the number of fertile generations (Chin et al, 1999), but eventually the germline cells from telomerase/p53 deficient mice also undergo apoptosis. This observation indicates that p53-independent mechanisms are involved in monitoring telomere integrity and genome instability in dividing cells (Chin et al, 1999). Tissue defects are related not to overall short telomere lengths per se, but to the inability to repair critically eroded telomeric tracts.
Telomere shortening has been linked to human syndromes with early mortality. For example, mutations in the telomerase components, TERT, TERC, DKC, NHP2, and NOP10 (and also in a shelterin component, TIN2—see below) are associated with rare human genetic disorders, including dyskeratosis congenita, aplastic anemia, and idiopathic pulmonary fibrosis (reviewed in Armanios, 2009; Calado and Young, 2009; Savage et al, 2009). In all but a few instances, these diseases are haploinsufficiency disorders, and 1 inactive allele is sufficient to confer disease. Partial loss-of-function of telomerase activity also leads to various forms of hematopoietic cancer. Similar disease phenotypes are recapitulated in murine models in which 1 or both copies of telomerase subunits or telomere-associated proteins have been deleted or mutated, alone or in combination with factors that promote genome instability (reviewed in Liu and Harrington, 2012).
Short telomeres have been observed in the peripheral blood of people with cardiovascular disease and those exposed to stress (Epel et al, 2006, 2010). Although a causal role of short telomeres has not yet been established, these observations suggest that telomere length could be a useful prognostic factor for certain lifestyle-associated diseases (Lin et al, 2011). In vitro, enforced overexpression of telomerase leads to telomere extension and can extend the life span of cells derived from people with dyskeratosis congenita harboring mutations in the telomerase RNA (Agarwal et al, 2011), and transcriptional activation of hTERT extends the replicative capacity of cells derived from HIV-infected patients (Fauce et al, 2008). Thus, telomere elongation could serve a useful therapeutic purpose in some human diseases.
5.7.3 Shelterin and Telomere Integrity
In mammals, telomeres are bound by a 6-subunit complex called shelterin, which contains the telomere-binding proteins TRF1, TRF2, and POT1, and their associated proteins RAP1, TPP1, and TIN2 (reviewed in de Lange, 2005; see Fig. 5–14). TRF1 and TRF2 bind to duplex telomeric DNA and anchor shelterin along the telomere repeats; POT1 binds to the single-stranded G-rich DNA overhang at telomeres; TIN2 is a protein scaffold for TRF1 and TRF2 and recruits POT1 to the complex via TPP1; and RAP1 associates with shelterin via binding to TRF2. Shelterin serves several regulatory roles at the telomere, including telomerase access to the telomere, telomere replication and integrity, t-loop assembly and stability, and the ability to attenuate ATM-dependent and ATR-dependent DNA damage responses (reviewed in de Lange, 2005). Shelterin function is conserved across a wide number of species, even in Drosophila where telomeres are maintained via a telomerase-independent mechanism of retrotransposition (Cenci et al, 2005; Longhese, 2008).
5.7.4 Telomere Maintenance and Cancer
To support long-term proliferative potential, cancer cells express telomerase activity or, more rarely, maintain telomeres via ALT. Evidence that telomerase inhibition selectively kills cancer cells was provided by studies showing that overexpression of dominant interfering variants of hTERT elicit telomere erosion and apoptosis in several cancer cell lines (Hahn et al, 1999; Herbert et al, 1999; Zhang et al, 1999). Several other lines of evidence now indicate that tumor cells must maintain minimally functional telomere reserves in order to retain proliferative potential.
In approximately 15% of cancers (across a wide variety of cancer types), cells maintain telomeres via HR between chromosome ends (reviewed in Cesare and Reddel, 2010) to employ “alternative lengthening of telomeres” (ALT). ALT cells possess promyelocytic leukemia-like nuclear bodies called APBs (ALT-associated PML bodies) that contain telomeric DNA, telomere DNA binding proteins, and DNA repair proteins and may represent sites permissive for telomere recombination. ALT cells also possess long and heterogenous telomere lengths, and carry extrachromosomal telomere DNA called t-circles (Cesare and Reddel, 2010). This latter feature may not be specific to ALT-associated cancers, as normal cells with elongated telomeres also exhibit t-circles (Pickett et al, 2011). In ALT cell lines, telomeres that are marked with a reporter construct can “jump” to new telomere locations, an observation that is consistent with direct telomere-telomere recombination (Dunham et al, 2000). Because ALT cells do not express telomerase activity, inhibition of telomerase function has no effect on their growth (Hahn et al, 1999).
As telomeres in actively dividing tumor cells are often short, and telomere lengths are longer in stem cells and other quiescent normal cells, telomerase is an attractive target for therapeutic intervention in cancer. In clinical development are several new telomere/telomerase targeting compounds including antisense oligonucleotides, G-quadruplex stabilizing substances, telomerase expression-related strategies such as telomerase promoter-driven suicide gene therapy, and telomerase immunotherapy (Harley, 2008). One such inhibitor, GRN163L is an antisense oligonucleotide directed against the RNA template of telomerase. This compound is now in Phase II clinical trials against several different types of cancer. An advantage of telom-erase inhibition is that it should be efficacious against all telom-erase-positive tumors, which make up more than 85% of tumor types. Compounds that disrupt telomere structure are also in development, and have the potential to treat both telomerase-positive and ALT-derived tumors (Harley, 2008). Inhibition of telomerase also holds promise for pediatric cancers but is complicated by the fact that pediatric tumors often possess longer initial telomere lengths (Tabori and Dome, 2007).
An important question is the role of critically short telomeres in the process of driving malignant transformation. For example, BRCA2 and BRCA1 are known to participate in processes that regulate telomere integrity, and patients harboring BRCA mutations show evidence of critically short telomeres and chromosome rearrangements that are consistent with loss of telomere integrity (Bodvarsdottir et al, 2011; Bogliolo et al, 2002; McPherson et al, 2006; Wang et al, 2007). The introduction of inducible chromosome breakage sites in murine ES cells has established that chromosome breakage within telomere DNA can contribute to the breakage-fusion-bridge cycles first noted in maize (Murnane and Sabatier, 2004). In prostate cancer short telomeres are observed in precursor lesions (ie high-grade prostatic intraepithelial neoplasia, or PIN) concomitant with the onset of other chromosomal rearrangements (Vukovic et al, 2003, 2007). Experiments in mice also suggest that, in some backgrounds predisposed to cancer, loss of telomere DNA can sometimes paradoxically lead to an increase in malignancy (Artandi and DePinho, 2010). Adjunct therapies that combine standard chemotherapeutic agents together with agents that perturb telomere function may be the best approach to achieve a rapid onset of apoptosis in telomerase-positive tumors and serve to mitigate the emergence of drug-resistant cancer.
5.8 DNA REPAIR AND CANCER TREATMENT
The PARP1 and PARP2 proteins are required for repair of DNA single-stranded breaks (SSBs) and in cells treated with small-molecule inhibitors of PARP (PARPi), unrepaired SSBs at replication forks are converted into DSBs, which require repair by HR to offset cell lethality. Synthetic lethality is the concept that mutation in 2 genes leads to death, whereas mutation of either alone is compatible with viability (see also Chap. 17). For example, tumor cells lacking HR function (eg, deficient in BRCA1 or BRCA2 expression) are exquisitely sensitive to PARPi because of the inability to repair replication-associated DSBs. Biomarkers that effectively predict functional losses in DNA repair activity, in addition to mutations in DNA repair genes, may therefore be useful in accurately predicting clinical PARPi efficacy (Dent and Bristow, 2011). Novel trials using PARPi may also be useful in patients who have abrogated function of the HR, FA, and PTEN pathways (Fraser et al, 2012). Similarly, deficiency in the MMR proteins MSH2 and MLH1 have been shown to be synthetically lethal with disruption of the DNA polymerases POLB and POLG, respectively (Martin et al, 2010), although clinically useful inhibitors of POLB or POLG are not yet available. A final example of synthetic lethality is the observation that FA defective cells are more sensitive to ATM inhibitors (Kennedy et al, 2007).
Documenting the functional loss or gain in DNA repair pathways may also be useful in sporadic tumors to individualize chemotherapy, radiotherapy, or molecular targeted therapies. Many human tumor types possess defects in DNA repair genes based on DNA, RNA, or protein analyses (Fig. 5–15), and these alterations can lead to differential sensitivity to DNA damaging agents. For example, a loss of function can lead to increased sensitivity to different chemotherapeutic agents or radiotherapy and elevated expression of the NER-associated Ercc1 gene is a promising biomarker for the relative resistance to cis-platinum chemotherapy in lung cancer (Cobo et al, 2007). In another example, glioblastoma brain cancer cells that express the enzyme called O-6-methylguanine-DNA methyltransferase (MGMT) are better able to repair the DNA damage caused by the alkylating agent temozolomide (Hegi et al, 2005), and the presence of MGMT protein in brain tumors predicts poor response to temozolomide. Epigenetic silencing of the MGMT/AGT gene can prevent the synthesis of this enzyme, and as a consequence such tumors are more sensitive to killing by temozolomide. Defects in HR can also sensitize tumors to agents that are highly toxic to HR-deficient tumor cells such as mitomycin C (MMC), cis-platinum and IR (Chan et al, 2010).
FIGURE 5–15 Diagram of DNA (green; mutation, deletion, or amplification), RNA (blue; under- or overexpression), or protein (black; under- or overexpression) alterations in DNA DSB response/repair proteins that have been identified in various tumor sites as indicated.
Related posts:
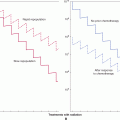
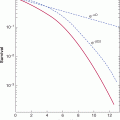
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


