Genetics of Type 2 Diabetes
Alessandro Doria
Type 2 diabetes, or non-insulin-dependent diabetes (NIDDM), is one of the most common metabolic disorders affecting humans. Approximately 14 million Americans have been diagnosed with this disease, and at least one third more are estimated to be affected without knowing it (1,2). Recent population surveys also indicate a striking increase in the prevalence of this disease in the United States, especially in younger people, with a 33% increase overall and a 70% increase in the 30- to 39-year age group during the 8-year period from 1990 to 1998 (2). Type 2 diabetes is now also a common diagnosis in the pediatric age group (3). The increasing number of affected people, together with the extensive list of long-term complications, including accelerated atherosclerosis, nephropathy, retinopathy, and neuropathy, makes type 2 diabetes a major health problem (4). The economic burden of diabetes is in excess of $100 billion annually in the United States alone. These factors have created a compelling need to understand the important role of genetic and environmental alterations in the etiology of this disease—to provide critical clues to the primary defects in this disorder, to allow tests that identify individuals at high risk of type 2 diabetes who may benefit from preventive programs, and to foster the development of new drugs to treat or prevent it.
GENES AND TYPE 2 DIABETES
Unlike type 1 diabetes, which is due to a single pathophysiologic defect (autoimmune destruction of β-cells resulting in a lack of endogenous insulin), most patients with type 2 diabetes exhibit two apparently different defects: (a) an impairment in the ability of muscle and fat to respond to insulin with increased glucose uptake and of liver to respond with decreased glucose output, i.e., insulin resistance; and (b) a failure of the β-cell to compensate for this insulin resistance by appropriately increasing insulin secretion (5) (Fig. 22.1). Although there are notable exceptions, in most longitudinal studies, insulin resistance can be demonstrated early in life before any evidence of glucose intolerance, whereas the β-cell failure develops somewhat later, in association with impaired glucose tolerance (5,6,7,8).
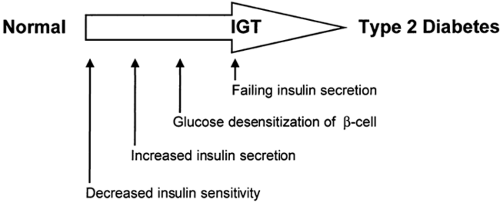 Figure 22.1. Model of the progressive pathogenesis of type 2 diabetes. In most individuals, there is a slow progression from normal or impaired glucose tolerance (IGT) to overt diabetes. This depends on interaction between genetic and environmental factors that regulate insulinsensitivity or the ability of β-cell to secrete insulin. |
The contribution of genetic factors to the development of insulin resistance, impaired insulin secretion, and type 2 diabetes has been known for many years (9,10). Supporting evidence includes the familial clustering of these traits (9,10,11), the higher concordance rate of type 2 diabetes in monozygotic versus dizygotic twins (12,13,14), and the high prevalence of type 2 diabetes in certain ethnic groups (e.g., Pima Indians or Mexican Americans) (8,15) (see Chapter 20 for further discussion of this topic). On the other hand, environmental factors also appear to play a role, as indicated by the increasing incidence of type 2 diabetes during the past decade, the well-known links to diet and lifestyle, and the differences in risk of type 2 diabetes among genetically similar populations living in different areas (2,16). Thus, diabetes can be viewed as a complex disorder, with genetic factors conferring an increased susceptibility upon which environmental factors must act in order for hyperglycemia to develop (Fig. 22.2). This model is analogous to those proposed for other multifactorial disorders such as cancer and hypertension (17). It is estimated that between 25% and 70% of the occurrence of type 2 diabetes can be attributed to genetic factors (14).
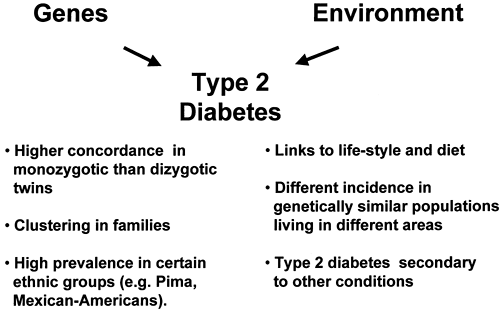 Figure 22.2. Contribution of genes and environment to the pathogenesis of type 2 diabetes. Available evidence supporting a role of genetic factors is listed on the left; the evidence for environmental factors is on the right. |
Studies of the patterns of inheritance indicate that multiple genes probably are involved, although their number and relative contributions are uncertain (18,19). As schematically illustrated in Fig. 22.3, different loci may contribute to the etiology of type 2 diabetes by interacting with each other within the same causal complex (epistasis). However, genes may also belong to different causal pathways, each being responsible for the development of diabetes in different subsets of individuals
(genetic heterogeneity). Some loci may be necessary but not sufficient (e.g., locus A in Fig. 22.3), whereas others may be neither necessary nor sufficient for the development of diabetes (e.g., loci B, C, and D). In this context, certain genes may predominate, their effects being more important than others (major gene effects). Some genetic characteristics may act only if patients are in the homozygous state (recessive inheritance), while for others the presence of only one allele may suffice (dominant inheritance). Furthermore, genetic susceptibility and environmental factors may interact in several different ways. For instance, environmental factors, such as excess caloric intake or sedentary lifestyle, may be responsible for the onset (initiation) of the metabolic abnormalities or β-cell damage, while genetic factors may be involved in regulating the rate of progression to overt diabetes. In other cases, genetic characteristics may be necessary from the very beginning for environmental factors to initiate the cascade of events leading to diabetes. Some of these genes may act at the level of insulin action while others may be involved in the regulation of insulin secretion.
(genetic heterogeneity). Some loci may be necessary but not sufficient (e.g., locus A in Fig. 22.3), whereas others may be neither necessary nor sufficient for the development of diabetes (e.g., loci B, C, and D). In this context, certain genes may predominate, their effects being more important than others (major gene effects). Some genetic characteristics may act only if patients are in the homozygous state (recessive inheritance), while for others the presence of only one allele may suffice (dominant inheritance). Furthermore, genetic susceptibility and environmental factors may interact in several different ways. For instance, environmental factors, such as excess caloric intake or sedentary lifestyle, may be responsible for the onset (initiation) of the metabolic abnormalities or β-cell damage, while genetic factors may be involved in regulating the rate of progression to overt diabetes. In other cases, genetic characteristics may be necessary from the very beginning for environmental factors to initiate the cascade of events leading to diabetes. Some of these genes may act at the level of insulin action while others may be involved in the regulation of insulin secretion.
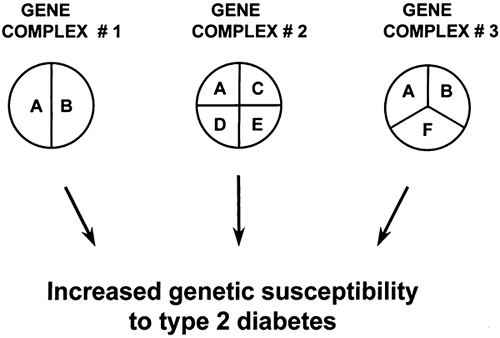 Figure 22.3. Illustration of the complex genetics of type 2 diabetes. Multiple type 2 diabetes genes (indicated by different letters of the alphabet) interact one with each other within different causal complexes that all lead to increased genetic susceptibility to the disease. |
This complexity makes the task of finding the genes for type 2 diabetes a formidable one. For instance, because of the important role played by environmental factors, cases of type 2 diabetes that are due entirely to nongenetic causes (so-called phenocopies) may be relatively frequent, especially in older populations. Conversely, diabetes may not develop in a substantial proportion of individuals carrying a susceptible genetic background because they have not been exposed to a diabetogenic environment or because they are not old enough (so-called nonpenetrants). Under these circumstances, research approaches that have been successful in identifying genes for mendelian disorders are difficult and of limited power. These problems, however, are counterbalanced by several major developments that have occurred during the past decade, namely the development of the polymerase chain reaction (20), the discovery of the class of DNA polymorphisms known as microsatellites and their systematic characterization as markers (21), the construction of genetic and physical maps of the human genome (22), the development of nonparametric methods of analysis of genetic data (23), and, finally, the completion of the Human Genome Project, which has provided the full human genome sequence and an initial catalogue of human genetic variation (24,25). The availability of these new tools carries with it the promise of finding the genes responsible for complex disorders, including type 2 diabetes.
THE SEARCH FOR TYPE 2 DIABETES GENES
A powerful approach to the identification of genetic variants that predispose to type 2 diabetes is to investigate their association with the disease in human populations (26,27). In their simplest version, these studies compare the distribution of the al-leles of a polymorphic locus between unrelated cases with type 2 diabetes and nondiabetic controls representative of the population from which cases have arisen (26). Any significant difference in allele distribution between cases and controls can be considered as evidence that this polymorphism or one associated (i.e., in linkage disequilibrium) with it contributes to the susceptibility to type 2 diabetes. Being case-control studies, association studies can be analyzed with the standard epidemiologic tools for evaluating disease risk according to various exposures, so that the presence of interaction between two loci or between a locus and environmental exposures can easily be tested. While straightforward in concept, association studies must be de-signed and analyzed according to established epidemiologic principles (26). False-positive results may arise due to unrecognized population differences between cases and controls (population stratification), while false-negative results can result from inappropriate study design such as inadequate sample size or inappropriate selection of cases and controls. The detection of small genetic effects may require several hundreds of cases and controls, especially if the linkage disequilibrium between the marker and the candidate susceptibility locus is weak (26,27). Despite these potential problems, association studies have nevertheless proved to be useful tools for detecting genetic contributions to human disorders, including diabetes. This is best exemplified by the case of HLA and type 1 diabetes (28).
It is estimated that about 11 million common variants (polymorphisms)-about 1 every 300 base pairs (bp)—are present along the human genome (29). Different combinations of these sequence differences account for heritable variation among individuals, including susceptibility to type 2 diabetes. Despite the remarkable progress in biotechnology, testing all 11 million variants for association with type 2 diabetes is not a feasible task at this time. To circumvent this problem, it has been proposed that the analysis be limited to a subset of polymorphisms that are spaced at regular intervals along the genome and serve as markers for intervening variants in linkage disequilibrium with these. The Human Genome Project has generated an extensive map of single-nucleotide polymorphisms (SNPs) that can be used for this task (25), but the number of markers needed to cover the whole genome may be disproportionally high [up to 500,000 according to one simulation (30)]. Another issue of concern is the stochastic variance of linkage disequilibrium throughout the genome, making the prospect of an effective whole-genome screen through this approach uncertain (31,32). Because of these constraints, association studies have thus far been focused on specific candidate genes selected according to two different strategies: (a) the study of genes that have been demonstrated to have a role in glucose homeostasis (functional candidate genes), and (b) the investigation of genes placed in chromosomal regions that are found to be inherited together (linked) with type 2 diabetes in whole-genome screens of families (positional candidate genes) (33). Such genome scans for linkage are not as sensitive as association studies, especially in the case of multifactorial disorders, but have the great advantage of not being limited to genes that are known to play a role in glucose metabolism (27). Both strategies have been widely used during the past decade and have led to the identification of many potential genes for type 2 diabetes (34). The most important of these loci, along with their positions along the genome, are reported in Figure. 22.4.
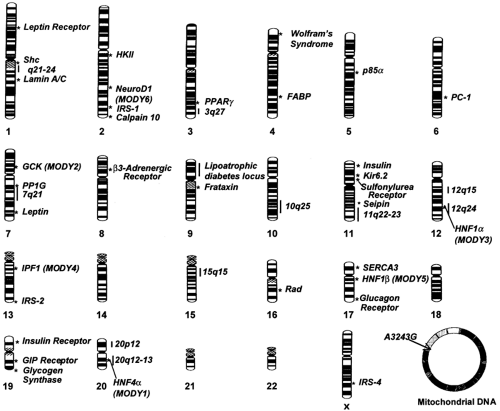 Figure 22.4. Location of potential type 2 diabetes genes in the human genome. (Modified from Almind K, Doria A, Kahn CR. Putting the genes for type II diabetes on the map. Nat Med 2001;7:277–279.) |
MONOGENIC FORMS OF DIABETES
The search has been particularly successful for special types of familial diabetes that are transmitted with a mendelian mode of inheritance and share some clinical features with type 2 diabetes. While these syndromes are fairly rare, their study has provided insights into the etiology of more common forms of type 2 diabetes and has led to the identification of new classes of candidate genes. Three main categories of monogenic forms of diabetes have been identified to date: maturity-onset diabetes of the young (MODY), genetic syndromes of extreme insulin resistance, and mitochondrial diabetes.
Maturity-Onset Diabetes of the Young
The term MODY was first introduced by Tattersall and Fajans in 1975 to indicate type 2–like diabetes with early onset (before age 25) and an autosomal dominant mode of inheritance (35). It is now clear that forms of autosomal dominant diabetes can occur after age 25 and that the inheritance pattern is more important than age at onset for the diagnosis (36,37,38). Autosomal dominant diabetes probably accounts for 1% to 3% of all cases of diabetes, although precise estimates of its prevalence are not available. The presence of large families with multiple affected members, coupled with the simple pattern of inheritance, has facilitated genetic studies of this disorder. Six different forms of MODY have been identified to date, each involving a mutation in a different gene located on a different chromosome (Table 22.1).
TABLE 22.1. Genes Found to Be Mutated in Autosomal Dominant Diabetes or Maturity-Onset Diabetes of the Young | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
The first MODY gene to be recognized was glucokinase (GCK1) (39,40,41,42), followed by hepatocyte nuclear factors (HNF) 1α and 4α (43,44). Glucokinase is the first enzyme involved in glucose metabolism in the β-cell (45), whereas HNF1α and HNF4α are transcription factors that regulate the expression of several genes in the liver and the β-cell (46,47). Glucokinase mutations are present in greater than 50% of French patients with MODY but are less frequent in other populations, representing
10% to 25% of cases in the United States and the United Kingdom and only 1% of cases in Japan (48). Mutations in HNF1α and HNF4α account for approximately 70% of cases of MODY in the United States and the United Kingdom (48,49). Rarer forms of MODY have also been found associated with mutations in other three transcription factors expressed in the liver and the β-cell, namely insulin promoter factor 1 (IPF1, also known as PDX1, IDX1, or STF1), HNF1β, and neuroD1 (also known as BETA2) (37,38,50). MODY due to mutations in IPF1 has been described in a single family, in which the homozygous proband had pancreatic agenesis and the heterozygous parents had diabetes (51). Diabetes due to mutations in HNF1α is also very rare and often associated with kidney cystic disease, genital malformations, or both (52,53). Two mutations in neuroD1 have been described, one in a typical MODY pedigree, the other in a family with “typical” type 2 diabetes, but with an autosomal dominant mode of inheritance (38).
10% to 25% of cases in the United States and the United Kingdom and only 1% of cases in Japan (48). Mutations in HNF1α and HNF4α account for approximately 70% of cases of MODY in the United States and the United Kingdom (48,49). Rarer forms of MODY have also been found associated with mutations in other three transcription factors expressed in the liver and the β-cell, namely insulin promoter factor 1 (IPF1, also known as PDX1, IDX1, or STF1), HNF1β, and neuroD1 (also known as BETA2) (37,38,50). MODY due to mutations in IPF1 has been described in a single family, in which the homozygous proband had pancreatic agenesis and the heterozygous parents had diabetes (51). Diabetes due to mutations in HNF1α is also very rare and often associated with kidney cystic disease, genital malformations, or both (52,53). Two mutations in neuroD1 have been described, one in a typical MODY pedigree, the other in a family with “typical” type 2 diabetes, but with an autosomal dominant mode of inheritance (38).
A prominent feature of all forms of MODY is a defect of insulin secretion (54,55). In the case of glucokinase mutations, this is due to an impairment of the β-cell glucose sensor caused by a decrease in the ability of glucokinase to phosphor-ylate glucose, resulting in inappropriately low fasting concentrations of insulin in relation to glycemic levels (45). For mutations in transcription factors, the cause of diabetes is less clear. One hypothesis is that a reduced activity of these transcription factors may impair the differentiation of islet cells during development, thereby reducing the number of functional β-cells in affected individuals (56,57,58,59). Another possibility is that these mutations directly affect the expression of genes required for glucose transport and metabolism in the β-cell (60). Also a direct effect on the expression of the insulin gene cannot be ruled out, especially for IPF1 and neuroD1, which are known regulators of insulin gene transcription (61,62). A clarification is expected from the postmortem analysis of islets from mutation carriers and from expression profile studies aimed at identifying the target genes of these transcription factors.
From a clinical standpoint, mutations in the glucokinase gene cause a rather mild form of diabetes (Table 22.1). Hyperglycemia is often diagnosed early in life, but fasting blood glucose values rarely exceed 130 mg/dL and the occurrence of diabetic complications is rare (54,63). Diabetes associated with mutations in HNF1α, HNF4α, and HNF1β is generally more severe (Table 22.1) (55). The insulin response to a glucose load is severely impaired, with insulin levels during an oral glucose tolerance test (OGTT) rarely exceeding 20 to 30 μU/mL (36,55). About 50% of diabetic carriers need insulin therapy, and diabetic complications are more common than in MODY due to glucokinase mutations (55,63). Diabetes due to IPF1 mutations is milder and is diagnosed at a slightly older age than is MODY due to HNF mutations (37). Of the two families thus far described as carrying neuroD1 mutations (38), one has a phenotype resembling that of MODY due to HNF1α mutations (young age at diagnosis, low prevalence of obesity, low insulin levels), while the phenotype in the other is more similar to that of common type 2 diabetes (older age at diagnosis, high prevalence of obesity, conserved insulin secretion).
While these are the general features of diabetes associated with different MODY genes, there is wide variability in the clinical presentation within the same MODY subtype. Some of this variability may be accounted for by the different nature of mutations segregating with diabetes in different families (allelic heterogeneity). However, large variation in the age at onset and severity of diabetes also is observed among carriers of identical mutations. This variability may be due to the presence of environmental or genetic factors that act as modifiers of the phenotype associated with each mutation. For instance, it has been hypothesized that co-inheritance of common type 2 diabetes susceptibility genes may determine a more severe presentation of MODY (64). Thus, MODY might not be the simple, monogenic disorder that has usually been considered, and further study of known MODY forms may lead to the discovery of new loci playing important roles in glucose homeo-stasis.
Finally, the six genes thus far identified do not account for all cases of autosomal dominant diabetes. In France and England, approximately 25% of MODY pedigrees do not show linkage with glucokinase, HNF1α, or HNF4α (48,65). The proportion of cases not accounted for by known genes appears to be even higher among families with an older age at diagnosis of diabetes (36). Available data suggest that some of these families are characterized by a pure insulin secretion defect, similar to that of HNF1α diabetes, whereas others are predominantly insulin-resistant, similar to common type 2 diabetes (36) (Fig. 22.5). Thus, a high degree of genetic heterogeneity is expected. Recent data from the laboratories of the Joslin Diabetes Center indicate that a locus accounting for diabetes in some of these families may be placed on chromosome 12q, 50 cM centromeric to HNF1α (66).
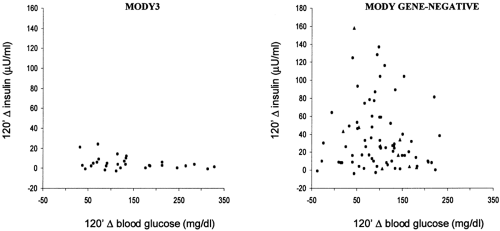 Figure 22.5. Relationship between increments in insulin secretion and blood glucose during oral glucose tolerance test (OGTT; 120′ Δ, change 2 hours after glucose ingestion) in maturity-onset diabetes of the young (MODY). Data are reported on the left for diabetic carriers of HNF1α mutations (MODY3) and on the right for diabetic members of families with early-onset, autosomal dominant type 2 diabetes not accounted for by known MODY genes (MODY gene-negative). Only individuals not treated with insulin were considered. MODY 3 subjects are characterized by a pure insulin secretion defect, while a more heterogeneous pattern is visible among MODY gene-negative subjects. (Copyright © 1999. From Doria A, Yang Y, Malecki M, et al. Phenotypic characteristics of early-onset autosomal-dominant type 2 diabetes unlinked to known maturity-onset diabetes of the young (MODY) genes. Diabetes Care 1999;22:253–261, with permission from the American Diabetes Association.) |
Genetic Syndromes of Extreme Insulin Resistance
Although rarer than MODY, syndromes of severe insulin resistance have been known for several years. Forms that have been described include leprechaunism, Rabson-Mendenhall syndrome, type A and type B syndromes, and lipodystrophies (67,68,69,70,71). Leprechaunism is a congenital syndrome characterized by severe insulin resistance of peripheral tissues, intrauterine and neonatal growth retardation, elfin facies, and early death (67). The Rabson-Mendenhall syndrome becomes evident during childhood, with severe insulin resistance, abnormal nails and dentition, accelerated growth, precocious pseudopuberty, and pineal hyperplasia (68). The type A syndrome of insulin resistance primarily affects young women, who present with severe hyperinsulinemia (>50 μU/mL in the fasting state), hyperandrogenism, polycystic ovary, and acanthosis nigricans (hyperpigmented and hyperkeratotic lesions in skinfold areas) (69). The type B syndrome has features similar to those of type A but affects older women and is often accompanied by autoimmune features (69). Finally, lipodystrophies are characterized by complete (lipoatrophic diabetes) or partial lack of adipose tissue, hepatosplenomegaly, hyperlipidemia, and severe insulin resistance, which can be present at birth or appear later in life (70,71).
Mutations in the insulin-receptor gene (INSR) impairing insulin action underlie all cases of leprechaunism and Rabson-Mendenhall syndrome described to date (72). Mutations in this gene are also responsible for a good proportion of cases of type A syndrome (72,73). Some of these sequence differences lead to a lower number of receptors because of decreased synthesis, increased degradation, or abnormal translocation from the endoplasm to the cell membrane (72). Other mutations are instead associated with decreased affinity of the receptor for insulin or impaired tyrosine kinase activity (72,73). In many instances, these forms are inherited in an autosomal recessive fashion, with some patients being homozygotes for a single mutation and others being compound heterozygotes who inherited two different mutant alleles, one from each parent. A few patients appear to have mutations in only one allele, suggesting an autosomal dominant pattern of inheritance, but the
presence of as yet unidentified mutations in the “healthy” allele cannot be excluded in these cases.
presence of as yet unidentified mutations in the “healthy” allele cannot be excluded in these cases.
Autosomal dominant forms of partial lipodystrophy (Dunnigan-Kobberling type) have recently been found to be due to mutations in the lamin A/C gene (LMNA) on chromosome 1q21 (74,75). Lamins are the main components of the nuclear lamina, the proteinaceous layer that is apposed to the inner nuclear membrane (76). The LMNA gene codes for two of these proteins, lamin A and C, through alternative splicing. Most of the mutations associated with partial lipodystrophy cluster on a 15-bp region of exon 8 of the LMNA gene and affect the globular C-terminal domain of the lamin A/C proteins (74,75). Especially frequent are substitutions of a highly conserved arginine at position 482. Little is known, however, about the mechanisms linking these mutations to lipodystrophy, insulin resistance, and hyperlipoproteinemia. Lamins are expressed in all tissues, where they play a role in DNA replication, chromatin organization, nuclear-pore arrangement, and anchorage of nuclear envelope proteins (76). Of note, mutations in the LMNA gene have also been found to be associated with Emery-Dreyfuss muscular dystrophy and dilated cardiomyopathy and conduction-system disease (77,78). In contrast with partial lipodystrophy, mutations in these disorders are distributed across several exons. Thus, the region around residue 482 in the globular domain of lamins A/C may determine specific functions in the adipocyte, the impairment of which leads to lipodystrophy. Mutations in the LMNA gene, however, do not appear to be involved in the etiology of generalized lipodystrophy [lipo-atrophic diabetes or Berardinelli-Seip syndrome (BSCL)] (79). A locus linked with some of these forms (BSCL2) has recently been mapped to chromosome 11q13 and shown to correspond to the human homologue of the murine guanine nucleotide-binding protein (G protein), γ3-linked gene (GnG31g) (80). BSCL2 is most highly expressed in brain and testis and codes for a protein, called seipin, of unknown function. Another BSCL locus (BSCL1) has been mapped on 9q34 in BSCL2 -negative families, but the identity of the gene that is involved is unknown (80).
Mitochondrial Diabetes
The mitochondrial genome consists of a circular 16,569-bp DNA molecule containing the genes for ribosomal and transfer RNAs and for some proteins of the respiratory chain. This DNA (mtDNA) has several unique features. First, it is transmitted only through the maternal side, since the sperm sheds its mitochondria during penetration into the ovum (81). Second, it exists in the cell in several copies—as many as a few thousand in cells with a high mitochondrial content. Finally, it is prone to a high degree of mutation and rearrangement, which, however, often affect only a fraction of the mtDNA, a phenomenon known as heteroplasmy. For each mutation, levels of heteroplasmy vary, being much higher in nondividing tissues such as muscle and brain than in rapidly dividing cells such as leukocytes.
A variety of syndromes associated with point mutations or deletions in the mtDNA have been described. Most of these concern tissues that have a high demand for energy, such as skeletal muscle and brain. The maternally inherited diabetes and neurosensory deafness (MIDD) syndrome is caused by a single nucleotide mutation (A3243G) in the mtDNA encoding the transfer RNA (tRNA) for leucine (82,83,84,85). Rare cases of MIDD have also been observed in association with mtDNA deletions or duplications (86). These mitochondrial defects are
thought to result in altered oxidative metabolism in the β-cell, leading to an impairment in the production of ATP that is needed for glucose-stimulated release of insulin (87,88). A reduction in the oxidative metabolism of glucose in peripheral tissues may also contribute to the development of hyperglycemia, but its role is uncertain. The MIDD syndrome accounts for approximately 1% of individuals with diabetes.
thought to result in altered oxidative metabolism in the β-cell, leading to an impairment in the production of ATP that is needed for glucose-stimulated release of insulin (87,88). A reduction in the oxidative metabolism of glucose in peripheral tissues may also contribute to the development of hyperglycemia, but its role is uncertain. The MIDD syndrome accounts for approximately 1% of individuals with diabetes.
Mitochondrial diabetes usually is characterized by an early onset of hyperglycemia (often before age 30), which is due to impaired insulin secretion and is followed by a rapid progression to insulin dependence (84,85). In some instances, MIDD can be misdiagnosed as type 1 diabetes (if insulin is required from the onset) or type 2 diabetes (if it can be managed with diet or oral medications), but patients are generally negative for islet-cell antibody and do not show increased prevalence of obesity (85). Impaired hearing, which can be so severe as to require the use of a hearing aid, is present in the majority of patients with MIDD and becomes manifest as a loss of high-frequency perception. The A3243G mutation also may lead to the so-called MELAS syndrome (mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes), which may or may not be associated with diabetes (89). Differences in phenotypic expression of the mitochondrial defects may be due to variations in the degree of heteroplasmy and to the tissue distribution of heteroplasmic mtDNA (85). The variable degree of heteroplasmy may also be responsible for difficulties in identifying the mitochondrial mutations responsible for the MIDD syndrome.
MULTIFACTORIAL FORMS OF TYPE 2 DIABETES
Because of the complex genetics, the search for genes involved in common, multifactorial type 2 diabetes has not been as successful. Many potential type 2 diabetes genes have been identified (Fig. 22.4), but findings have been difficult to replicate in most cases. Several factors contribute to these variable results. An important issue may be the presence of genetic heterogeneity, with different genes contributing to type 2 diabetes in different populations. Even within the same ethnic group, different subtypes of type 2 diabetes may be accounted for by different genes, making the results of association studies heavily dependent on how affected cases or families are selected. Discrepancies also can occur because of population differences in the linkage disequilibrium between markers and causal variants. Other important factors concern methodologic issues regarding the study design and the analysis of data. False-positive results can arise from multiple hypothesis testing or, in association studies, from unrecognized ethnic differences between cases and controls (so-called population stratification). Conversely, studies may fail to replicate true positive results because of inadequate statistical power or of failure to account for gene-gene or gene-environment interactions. All these factors must be carefully considered when weighing the evidence supporting or excluding the role of a certain locus. As discussed above, two main approaches have been followed to identify genes for multifactorial forms of type 2 diabetes: (a) studies of functional candidate genes and (b) whole-genome screens followed by the study of positional candidate genes.
Studies of Functional Candidate Genes
Selection of this class of candidate genes for type 2 diabetes has been based upon the biochemical function and expression pattern of the encoded proteins, which, if altered, might be implicated in the pathogenesis of the disease. Since both insulin resistance and a deficit of insulin production are involved in the etiology of type 2 diabetes (5), genes known to be involved in insulin action or β-cell function have been intensely investigated. Because of the strong links between obesity and type 2 diabetes, many studies have also considered genes involved in the regulation of the energy balance and in the development and metabolism of adipose tissue.
GENES INVOLVED IN INSULIN ACTION
Insulin action at the cellular level is the result of a complex network of signaling events (90) (Fig. 22.6). Binding of insulin to its
receptor leads to activation of the insulin receptor tyrosine kinase, which results in phosphorylation of tyrosine residues on a family of insulin receptor substrates. These tyrosine-phosphor-ylated substrates then bind to and activate a number of intracellular proteins in the insulin signaling cascade, the most important of which for metabolic effects is phosphatidylinositol (PI) 3-kinase. Docking of these proteins in turn activates specific signal transduction pathways (for instance, the translocation of glucose transporters to the plasma membrane) mediating the cellular effects of insulin.
receptor leads to activation of the insulin receptor tyrosine kinase, which results in phosphorylation of tyrosine residues on a family of insulin receptor substrates. These tyrosine-phosphor-ylated substrates then bind to and activate a number of intracellular proteins in the insulin signaling cascade, the most important of which for metabolic effects is phosphatidylinositol (PI) 3-kinase. Docking of these proteins in turn activates specific signal transduction pathways (for instance, the translocation of glucose transporters to the plasma membrane) mediating the cellular effects of insulin.
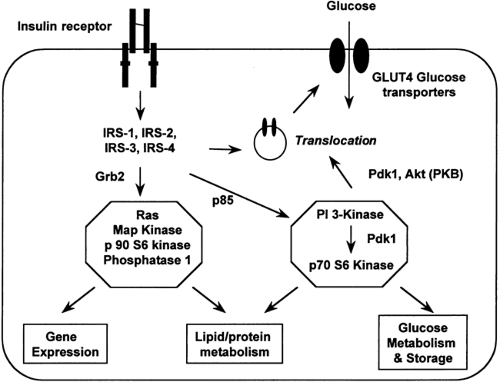 Figure 22.6. Schematic model of the cellular path-ways involved in insulin signaling. IRS, insulin-receptor substrate; PDK, phosphoinositide-dependent protein kinase; PKB, protein kinase B; Grb, growth factor receptor-bound protein; p 90 S6 kinase and p 70 S6 kinase, 90 kDa and 70 kDa ribosomal kinase, respectively; PI, phosphatidylinositol. |
Insulin Receptor
Despite the relatively high frequency of insulin-receptor mutations in syndromes of severe insulin resistance, genetic variability in the insulin receptor gene does not appear to play a major role in common forms of type 2 diabetes. Initial studies demonstrated several polymorphisms in the coding regions, some affecting the amino acid sequence, but none of these were shown to be more frequent in subjects with type 2 diabetes as compared with controls (91,92). Results were similarly negative in populations with a high prevalence of insulin resistance such as Pima Indians and in linkage analyses of families (93,94,95). More recently, a significant association has been reported between type 2 diabetes and the Val985Met variant in two Dutch populations (96). This polymorphism appears to confer a twofold increase in the risk of type 2 diabetes but, being rather rare, can account for only a handful of cases of type 2 diabetes. Since the INSR gene is rather large, noncoding regions have not been extensively screened for variation. Thus, one cannot rule out the existence of variants in as-yet unscreened regulatory elements that increase susceptibility to diabetes by impairing insulin-receptor expression. Indeed, a decrease in the number of insulin receptors is observed in type 2 diabetes, although this is common to all hyperinsulinemic states and is thought to be due to downregulation of the receptor through increased degradation (97). An additional factor is the alternative splicing of the messenger RNA, which originates two forms of the receptor differing by 12 amino acids near the C-terminus of the α-subunit (98,99). These two receptor isoforms exhibit subtle differences in binding affinities, tyrosine kinase activities, and internalization kinetics (100,101,102). Differences in the expression of these two subtypes between patients with type 2 diabetes and nondiabetic controls have been reported in the literature, but the significance of this result is controversial because not all studies have found this difference and the functional differences between the two isoforms are rather small (103,104,105). Whether the alternative splicing is under the control of genetic variants is unknown at this time.
Insulin-Receptor Substrates
One of the best-characterized molecules in the intracellular insulin action cascade is insulin-receptor substrate-1 (IRS-1), a 131-kilodalton (kDa) cytosolic protein that is tyrosine phosphorylated by the insulin receptor (106,107). In animal models, heterozygous disruption of IRS-1 does not lead to frank hyperglycemia but acts epistatically to potentiate other genetic defects in the production of diabetes (108,109). A total of nine amino acid substitutions have been identified in this molecule to date in white and Japanese populations (110,111,112,113,114,115,116,117,118,119,120,121,122). The most prevalent of these is a glycine-to-arginine substitution placed at codon 972 between two potential tyrosine phosphorylation sites binding the p85 subunit of PI 3-kinase (110). This variant has been examined for association with type 2 diabetes in more than 14 populations (110,111,112,113,114,115,116,117,118,119,120,121,122). While initial studies suggested an association with type 2 diabetes (110), the carrier frequency varies considerably among different populations (from 0.01 to 0.19) and an association with type 2 diabetes has not been observed in all populations (Fig. 22.7). If all these studies are considered together in a meta-analysis, the relative risk of type 2 diabetes associated with the Gly972Arg variant is not significantly different from 1 [odds ratio (OR): 1.05, 95% confidence interval (CI) 0.83–1.33]. Results are similar if the meta-analysis is performed separately for whites and Asians.
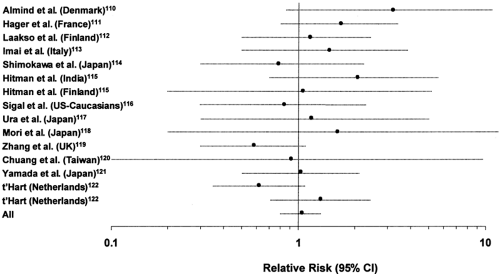 Figure 22.7. Estimated risk of type 2 diabetes associated with the IRS-1 Gly972Arg variant. For each study, the solid circles represent the odds ratio for 972Arg carriers and the dotted line indicates the 95% confidence interval. Odds ratios were not significantly different across studies (p = 0.50, Breslow-Day test). “All” indicates the Mantel-Haenszel estimate of the common relative risk based on all published studies (4,306 individuals). |
Despite these negative results, IRS-1 cannot be easily dismissed as a possible type 2 diabetes gene. All the studies above considered cases of type 2 diabetes that were not selected on the basis of patients’ insulin sensitivity or body weight. In contrast, it has been shown that the Gly972Arg is specifically associated with a subset of type 2 diabetes characterized by obesity and marked insulin resistance (115,123). Furthermore, functional data support a role for this variant in the etiology of insulin resistance. Transfection studies have consistently shown that the Arg972 allele is associated with a 40% decrease in IRS-1-associated PI 3-kinase activity and a 25% to 40% decrease in binding of the p85 subunit of PI 3-kinase to IRS-1 (124,125). It is interesting that expression of this variant in β-cells decreases both glucose and sulfonylurea-stimulated insulin secretion and that carriers of this allele have lower insulin-secretion rates during hyperglycemic clamp than do noncarriers (126,127). Thus, the Gly972Arg variant has the potential to contribute to both insulin resistance and the defective β-cell compensation characteristic of type 2 diabetes.
IRS-2 has been also at the center of intense investigation following the report that homozygous disruption of this gene in mice results in a phenotype similar to human type 2 diabetes (128). However, all three amino acid substitutions that have been identified in humans (Leu647Val, Gly879Ser, and Gly1057Asp) do not appear to be associated with type 2 diabetes, although nondiabetic Asp1057 homozygotes may have decreased insulin levels both in the fasting state and during OGTT (129,130,131). Variability in the coding region of the IRS-4 gene (placed on chromosome X) has also been excluded as a possible genetic determinant of type 2 diabetes (132). No information is available on the role of IRS-3, as the gene for this protein has been identified in mouse and rat but not in humans.
PI 3-Kinase
PI 3-kinase is the major protein that is bound and activated by phosphorylated IRSs (90). This molecule phosphorylates the inositol ring of phosphatidylinositol, producing phosphatidylinositol 4-phosphate (P), -4,5-P2, and 3,4,5-P3, which propagate the insulin signal to downstream pathways (133). A reduction in the insulin response of this enzymatic activity has been reported in muscle and adipose tissue from insulin-resistant subjects with type 2 diabetes (134). The enzyme consists of a catalytic subunit (p110) coupled to a regulatory subunit (p85) having two isoforms (α and β) encoded by different genes (135). A third regulatory subunit with lower molecular weight has also been isolated (p55γ) along with low-molecular-weight splice variants of p85α (136). A relatively common amino acid substitution (Met326Ile) has been identified in the p85α isoform (137). This residue is highly conserved across species and is placed in proximity to one of the two SH2 domains that interact with phosphorylated IRSs. While the Ile326 allele is not associated with type 2 diabetes, it was correlated with reductions in whole-body glucose effectiveness and intravenous glucose disappearance in healthy Danish subjects (137). In sharp contrast with these findings, the variant had an “antidiabetogenic” effect in Pima women, as indicated by a significantly higher acute insulin response during OGTT in Ile/Ile homozygotes (138). No effect was observed in Japanese patients (139). Results of in vitro experiments have failed to show significant difference in insulin-stimulated lipid kinase activity and phosphotyrosine recruitment between Met326 and Ile326 allele (140). Thus, if this polymorphism affects insulin action, its effect is probably minor. A rare variant of p85α (Arg409Gln) has been described in one subject with extreme insulin resistance and appears to be associated with lower insulin-stimulated PI 3-kinase activity (140). A screening of the p110 catalytic unit in Finnish subjects has identified two polymorphisms in the 5′ flanking region, but these are not associated with diabetes (141). The effect of genetic variability in the p85β and p55γ isoforms has not been investigated to date.
Genes Involved in Glucose Transport and Metabolism
While insulin has pleiotropic effects on cellular functions, the actions that have been specifically shown to be impaired in type 2 diabetes are those related to glucose uptake and incorporation into glycogen (5). In muscle and fat cells, insulin stimulates glucose uptake by inducing the translocation of glucose transporter 4 (GLUT4) from intracellular compartments to the cell surface and activates glycogen synthase by promoting its dephosphorylation (90). Both effects appear to involve the activation of the phosphoinositide-dependent protein kinase-1 (PDK1) and protein kinase B (PKB, also known as AKT) (Fig. 22.6), which have been both screened for amino acid variants in subjects with insulin resistance. However, only silent polymorphisms have been found (142). Likewise, variability in the GLUT4 gene (17p13) does not seem to contribute to type 2 diabetes. Sequence differences in GLUT4 promoter and coding sequence have been identified, but are either extremely rare or equally frequent in type 2 diabetic cases and nondiabetic controls (92,143,114,145).
The glycogen synthase gene (19q13) has been intensively investigated because of the finding of multiple defects in the activity of this enzyme in the skeletal muscle of patients with type 2 diabetes (146). An Xba I restriction fragment length polymorphism (RFLP) placed in intron 14 has been reported to be associated with type 2 diabetes, impaired nonoxidative glucose metabolism, and arterial hypertension in Finland (147). This finding has been replicated by the same authors in a different sample of unrelated Finnish individuals and in affected sib-pairs (148,149) but has not been confirmed in populations from France, Japan, and the United States (150,151, and A. S. Krolewski, personal communication). An association has been found in Pima Indians and Japanese but concerns a dinucleotide repeat rather than the Xba I polymorphism (152). A possible explanation is that both the Xba I polymorphism and the dinucleotide repeat are markers in linkage disequilibrium with as-yet-unidentified disease variants that are characteristic of specific populations. None of the amino acid polymorphisms identified to date appear to contribute to insulin-resistant phenotypes, although the very rare Pro442Ala variant has been shown to significantly decrease the ability of the enzyme to synthesize glycogen (153,154). A polymorphism in the regulatory G subunit of the glycogen synthase-activating protein phosphatase 1 (Asp905Tyr) may have a minor impact on insulin sensitivity in Danes, while a common variant in the 3′-untranslated region (UTR), causing a tenfold difference in reporter mRNA half-life, may contribute to insulin resistance in Pima Indians and aboriginal Canadians (155,156,157).
Inhibitors of Insulin Action
All the candidate genes described above have been selected based on the hypothesis that a genetically determined deficiency in elements of insulin action pathways leads to insulin resistance. An alternative concept is that insulin resistance may be due to a genetically determined excess in the expression or activity of inhibitors of insulin action. Several proteins that inhibit insulin signaling have been identified to date, and some of these have been investigated for a genetic contribution to type 2 diabetes (Fig. 22.8).
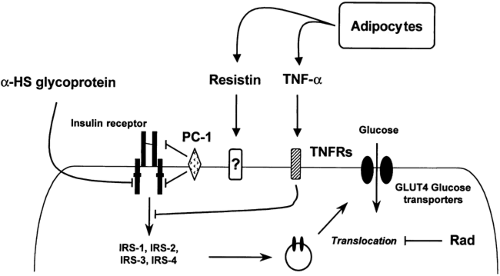 Figure 22.8. Natural inhibitors of insulin signaling and their action sites. TNF, tumor necrosis factor; TNFRs, TNF receptors; IRS, insulin-receptor substrate; PC, plasma cell membrane glycoprotein. |
Rad is a member of a unique family of Ras-related guanosine triphosphatases that was identified by subtraction cloning between skeletal muscle of a type 2 diabetic individual and a nondiabetic subject (158). Rad expression is
increased in about 20% of subjects with type 2 diabetes, its content being correlated to body weight and resting metabolic rate. The overexpression of this protein in cultured myocytes and adipocytes inhibits insulin-stimulated uptake of glucose through an effect on the intrinsic activity of GLUT4 molecules rather than on their translocation (159,160). The gene coding for Rad consists of 5 exons spanning about 3.7 kb on chromosome 16q (161). In a white population from the United States, a significant association was found between type 2 diabetes and the rare alleles of a composite trinucleotide repeat polymorphism placed in intron 2 (162). Carriers of these alleles had a threefold increase in the risk of diabetes as compared with noncarriers. This association, however, was not confirmed in Finns (163). By contrast, an association was detected in Japa-nese, but the alleles associated with type 2 diabetes are different from those found in whites in the United States (164). These discrepancies may reflect ethnic differences in the pattern of linkage disequilibrium between the trinucleotide repeat marker in exon 2 and a causal variant at the Rad locus or its vicinity.
increased in about 20% of subjects with type 2 diabetes, its content being correlated to body weight and resting metabolic rate. The overexpression of this protein in cultured myocytes and adipocytes inhibits insulin-stimulated uptake of glucose through an effect on the intrinsic activity of GLUT4 molecules rather than on their translocation (159,160). The gene coding for Rad consists of 5 exons spanning about 3.7 kb on chromosome 16q (161). In a white population from the United States, a significant association was found between type 2 diabetes and the rare alleles of a composite trinucleotide repeat polymorphism placed in intron 2 (162). Carriers of these alleles had a threefold increase in the risk of diabetes as compared with noncarriers. This association, however, was not confirmed in Finns (163). By contrast, an association was detected in Japa-nese, but the alleles associated with type 2 diabetes are different from those found in whites in the United States (164). These discrepancies may reflect ethnic differences in the pattern of linkage disequilibrium between the trinucleotide repeat marker in exon 2 and a causal variant at the Rad locus or its vicinity.
Another potential inhibitor of insulin action is the plasma cell membrane glycoprotein-1 (PC-1), a transmembrane protein with phosphodiesterase and pyrophosphatase activity (165). When overexpressed in cultured cells, PC-1 inhibits insulin receptor tyrosine kinase activity by a direct interaction with the insulin receptor α-subunit (166). In nonobese, nondiabetic subjects, the PC-1 content of skeletal muscle is inversely correlated to both in vivo insulin action and in vitro stimulation of the insulin receptor tyrosine kinase activity (167). An amino acid variant identified in PC-1 (Lys121Gln) has been reported to contribute to insulin resistance in a nondiabetic population from Sicily, primarily by impairing insulin receptor kinase activity (168). Gln121 carriers have a threefold increase in the risk of being hyperinsulinemic and insulin resistant but have no increase in risk of developing type 2 diabetes (168). Fibroblasts from these subjects show an impairment of insulin-receptor autophosphorylation in the presence of normal PC-1 levels. Similar findings of association have been obtained in a family-based study from Sweden but not in a large population-based study from Denmark (169,170). In Mexican Americans, a major locus for fasting insulin concentrations and insulin-resistance traits has recently been mapped to the same region of the PC-1 gene on chromosome 6q22–q23 (171). Whether this locus corresponds to the PC-1 gene in this population awaits further investigation.
Other studies have considered circulating inhibitors of insulin action. pp63, also known as α-HS glycoprotein, was identified in extracts of rat liver (172). In both rodents and humans, this protein is present at relatively high levels in the circulation, where it might act on peripheral tissues involved in insulin action. pp63 has been shown to inhibit the insulin receptor tyrosine kinase and insulin-stimulated DNA synthesis (173). Thus far, however, there is no evidence for genetic alterations in expression, secretion, or action of this protein in type 2 diabetes. A circulating inhibitor that has been more intensively studied is tumor necrosis factor-α (TNF-α), a potent cytokine with a wide range of proinflammatory activities (174). TNF-α may contribute to the insulin resistance of infection or stress and also to type 2 diabetes, since TNF-α is hypersecreted by adipocytes in obesity (175). Exposure to TNF-α decreases glucose uptake in vitro and in vivo, and homozygous disruption of the TNF-α gene protects mice from obesity-induced insulin resistance (175,176). However, whether genetic variability in this cytokine contributes to the development of type 2 diabetes in humans remains unclear. The human TNF-α gene lies in the class III region of the major histocompatibility complex, centromeric to the HLA-B locus and telomeric to HLA-DR. The rare allele of a polymorphism in the promoter region (G/A at position -308) is associated with increased TNF-α expression and more severe disease in infections such as malaria and leishmaniasis (177,178). The same polymorphism was found to be associated with indices of insulin resistance in a Spanish population but not in studies from the United States, Germany, Denmark, United Kingdom, Japan, and Hong Kong (179,180,181,182,183,184). Another promoter variant (G/A at -238) was reported to be associated with impaired insulin sensitivity in the United Kingdom, but this was not confirmed in other populations (179,180,181,182,183,184,185). The role of variability in the two TNF-α receptor (TNFR1 and TNFR2) has been less investigated. Only one report has been published to date describing lower insulin sensitivity in carriers of an allele in the 3′ UTR of TNFR2 (186).
Related posts:
 Genetic Regulation of Islet Function
Magnetic Resonance Spectroscopy Studies of Liver and Muscle Glycogen Metabolism in Humans
Epidemiology of Diabetes Mellitus
Diabetes in Minorities in the United States
Behavioral Research and Psychological Issues in Diabetes: Progress and Prospects
Pancreas and Islet Transplantation
Genetic Regulation of Islet Function
Magnetic Resonance Spectroscopy Studies of Liver and Muscle Glycogen Metabolism in Humans
Epidemiology of Diabetes Mellitus
Diabetes in Minorities in the United States
Behavioral Research and Psychological Issues in Diabetes: Progress and Prospects
Pancreas and Islet Transplantation
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


