(1)
Department of Surgery, Dar A lAlafia Medical Company, Qatif, Saudi Arabia
2.1 Introduction
The normal hemoglobin is made up of two parts, heme and protein. The protein is made up of four polypeptide chains (Fig. 2.1):
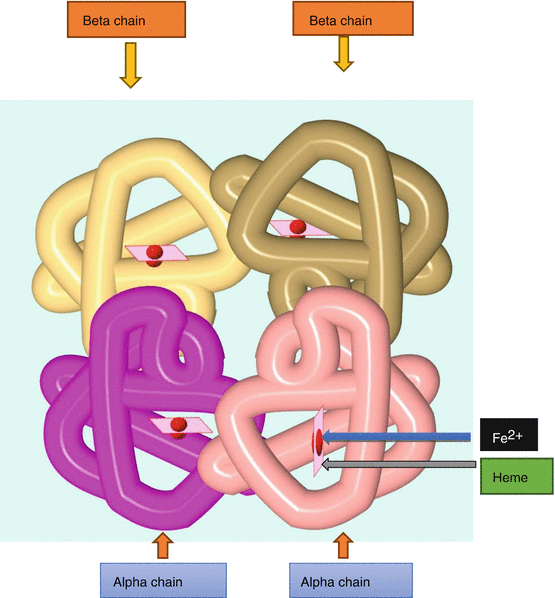
Fig. 2.1
Diagrammatic representation of the normal hemoglobin which is made up of heme and globin (2 alpha chains and 2 beta chains)
2 alpha chains (141 amino acids long)
2 beta chains (146 amino acids long)
There is a gene responsible for the alpha globin peptide chain and another gene for the beta globin peptide chain.
The HBB gene which is responsible for the beta globin chains is located on the short arm of chromosome 11.
There are many known mutations in the HBB gene. These lead to a variety of inherited diseases.
A mutation in the HBB gene produces abnormal versions of beta-globin such as hemoglobin C (HbC), hemoglobin E (HbE), and hemoglobin S (HbS). Sickle cell anemia is caused by a mutation in the HBB gene.
Sickle cell anemia is due to an autosomal recessive allele which is found on the short arm of chromosome 11.
The major sickle genotypes described so far include the following:
HbSS disease or sickle cell anemia (homozygous for the S globin): This is the most common form with usually severe or moderately severe symptoms.
HbS/b-0 thalassemia (double heterozygous for HbS and b-0 thalassemia): This is clinically indistinguishable from sickle cell anemia.
HbS/b + thalassemia: This is characterized by mild to moderate severity with variability in different ethnicities.
HbSC disease (double heterozygous for HbS and HbC): This is characterized by moderate clinical severity.
HbS/hereditary persistence of fetal Hb (S/HPHP): This is characterized by very mild or asymptomatic phenotype.
HbS/HbE syndrome: This is very rare with a phenotype usually similar to HbS/b + thalassemia.
Rare combinations of HbS with other abnormal hemoglobins such as HbD Los Angeles, G-Philadelphia, HbO Arab, and others.
In sickle cell anemia, the genetic code is altered leading to the substitution of a single amino acid where the amino acid glutamic acid is replaced by valine in the sixth position of the 146 amino acids of the beta chain of hemoglobin (Figs. 2.2, 2.3, 2.4, and 2.5).
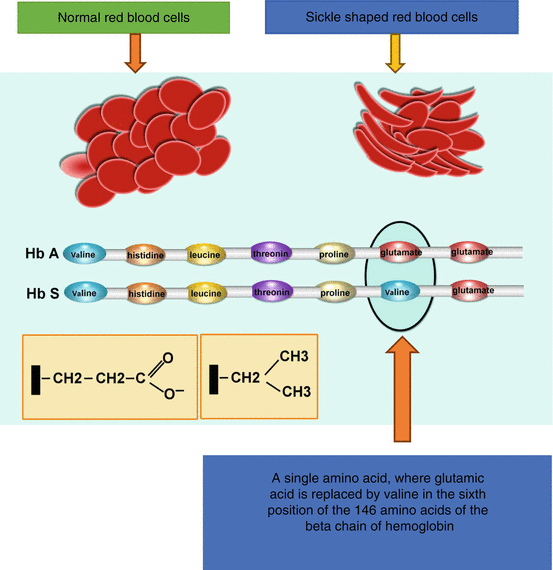
Fig. 2.2
Diagrammatic representation showing the change of a single amino acid, where glutamic acid is replaced by valine in the sixth position of the 146 amino acids of the beta chain of hemoglobin. This leads to the formation of hemoglobin S and a change of the normal red blood cells to sickle-shaped red blood cells
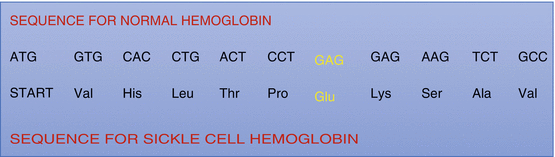
Fig. 2.3
Diagrammatic representation showing the amino acids sequence in the normal hemoglobin and sickle cell hemoglobin. The change is at the sixth position among the 146 amino acids of the beta chain of hemoglobin where glutamic acid is replaced by valine

Fig. 2.4
Diagrammatic representation showing the nucleotide and amino acids sequence in the normal hemoglobin and sickle cell hemoglobin. The change is at the sixth position among the 146 amino acids of the beta chain of hemoglobin where glutamic acid nucleotide (GAG) is replaced by valine nucleotide (GTG). This will lead to the replacement of valine instead of glutamic acid
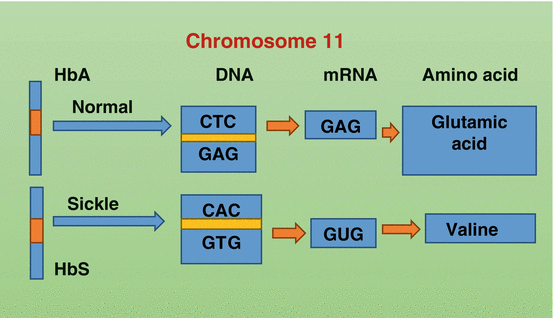
Fig. 2.5
Diagrammatic representation showing chromosome 11 and the changes in the nucleotide and amino acids sequence in the normal hemoglobin and sickle cell hemoglobin. The change will lead to the replacement of valine instead of glutamic acid
The mutation causing sickle cell anemia is a single nucleotide substitution (A to T) in the codon for amino acid 6. The change converts a glutamic acid codon (GAG) to a valine codon (GTG). As a result of this change, a modified hemoglobin is produced. This is referred to as HbS (Figs. 2.6, 2.7, and 2.8).
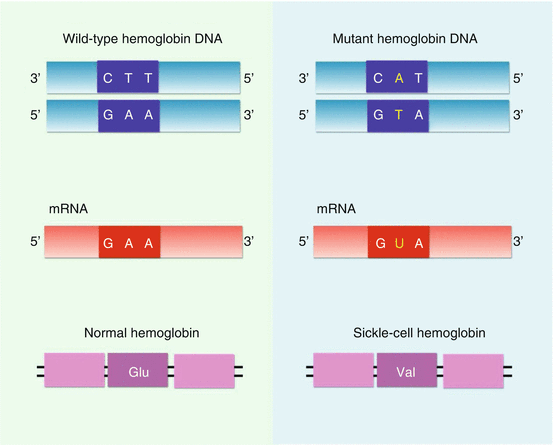
Fig. 2.6
Diagrammatic representation showing a single nucleotide substitution (A to T) in the codon for amino acid 6. Glutamic acid is coded by either GAA or GAG. Valine is coded by GUA, GUC, and GUU or GUG. In the mRNA, the change converts a glutamic acid codon (GAG or GAA) to a valine codon (GUA, GUC, GUU, or GUG)
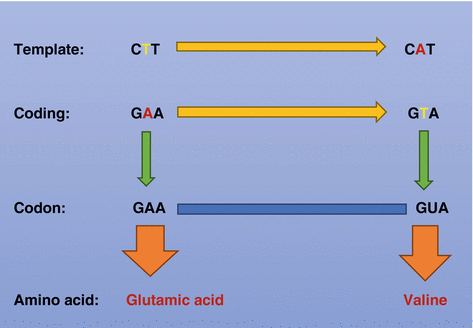
Fig. 2.7
Diagrammatic representation showing a single-nucleotide substitution (A to T) in the codon for amino acid 6 in patients with sickle cell anemia. The end result is a change of the codon for glutamic acid (GAA) to the codon for valine (GUA). This leads to the production of the amino acid valine instead of glutamic acid in the sixth position of the beta chain of hemoglobin
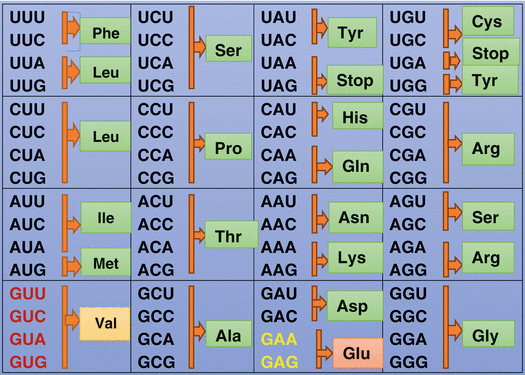
Fig. 2.8
Diagrammatic representation showing the codons for each amino acid. Glutamic acid is coded by either GAA or GAG. Valine is coded by GUA, GUC, and GUU or GUG. In patients with sickle cell anemia, there is a single change of one amino acid where glutamic acid at the sixth position among the 146 amino acid of the hemoglobin beta change is replaced by valine. This leads to a change and alteration of the hemoglobin and the hemoglobin tend to stick to each other, forming long insoluble fibers of hemoglobin within the red blood cell. The red blood cells become more fragile, adhering to each other and rupturing easily
HbS arises from a mutation substituting thymine for adenine in the sixth codon of the beta-chain gene, GAG to GTG. This causes coding of valine instead of glutamate in position 6 of the Hb beta chain. The resulting Hb has the physical properties of forming polymers under deoxy conditions. It also exhibits changes in solubility and molecular stability. These properties are responsible for the profound clinical expressions of the sickling syndromes.
This single change of one amino acid, valine for glutamic acid substitution, results in hemoglobin tetramers that aggregate into arrays upon deoxygenation in the tissues. This aggregation leads to deformation of the red blood cell, changing them into sickle-shaped cells and making them relatively inflexible and unable to traverse the capillary beds. Repeated cycles of oxygenation and deoxygenation lead to irreversible sickling (Figs. 2.9, 2.10, 2.11 and 2.12).
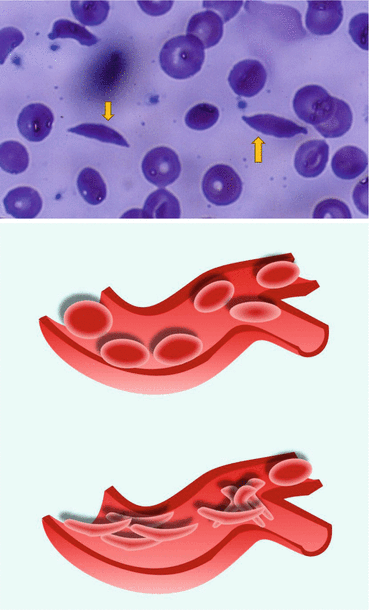
Figs. 2.9 and 2.10
A blood film showing sickled red blood cells and a diagrammatic representation of normal and sickled red blood cells causing occlusion of blood vessels
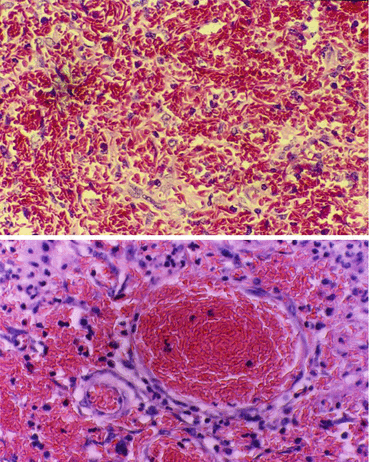
Figs. 2.11 and 2.12
A microscopic picture showing sickling of red blood cells and hemosiderosis. In the lower picture, note also the sickled cells within the blood vessel
As a result of this:
The hemoglobin molecule is altered so that it becomes more hydrophobic.
When the altered hemoglobin chains fold into their three-dimensional shape, they tend to stick to each other, forming long insoluble fibers of hemoglobin within the red blood cell.
The red blood cells are deformed by this altered hemoglobin. They become more fragile, rupturing easily and adhering to each other leading to closure of the small blood vessels.
Sickle cell anemia is an autosomal recessive disorder. Affected individuals must carry two copies (homozygous genotype = SS) of the HbS gene. Individuals who are heterozygous (genotype = AS) have what is referred to as sickle cell trait.
2.2 Risk of Inheritance
People inherit a pair of genes that regulate the production of hemoglobin.
One gene comes from each parent.
Sickle cell anemia:
This results from inheritance of two sickle genes, one from each parent.
Sickle cell trait:
This occurs if a person inherits one normal hemoglobin gene and one sickle cell gene.
People who have sickle cell trait are healthy, but they are “carriers” who can pass the disease on to their children.
They are also prone to develop sickle cell anemia like symptoms but under certain conditions.
The risk of a child inheriting sickle cell anemia or sickle cell trait is as follows:
If both parents have sickle cell trait (each has one normal hemoglobin gene and one sickle cell gene) (Fig. 2.13):
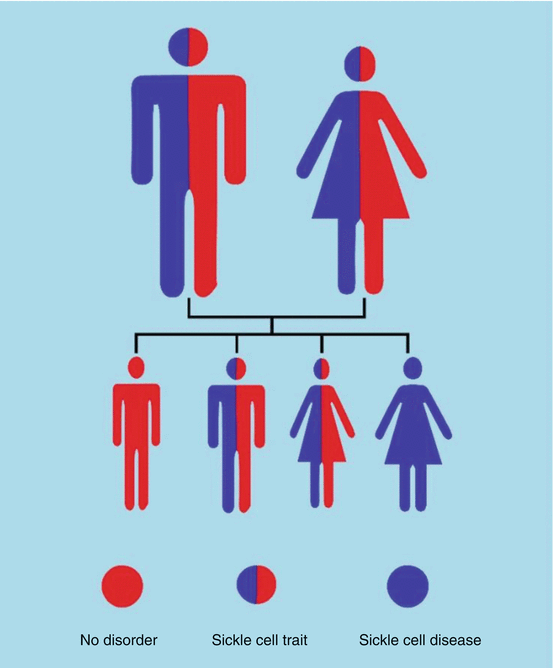
Fig. 2.13
Diagrammatic representation of inheritance if both parents have sickle cell trait
The child has a 50 % chance of inheriting sickle cell trait (one normal gene, one sickle cell gene).
The child has a 25 % chance of inheriting sickle cell anemia (two sickle cell genes).
The child has a 25 % chance of not inheriting either the trait or the disease (two normal genes).
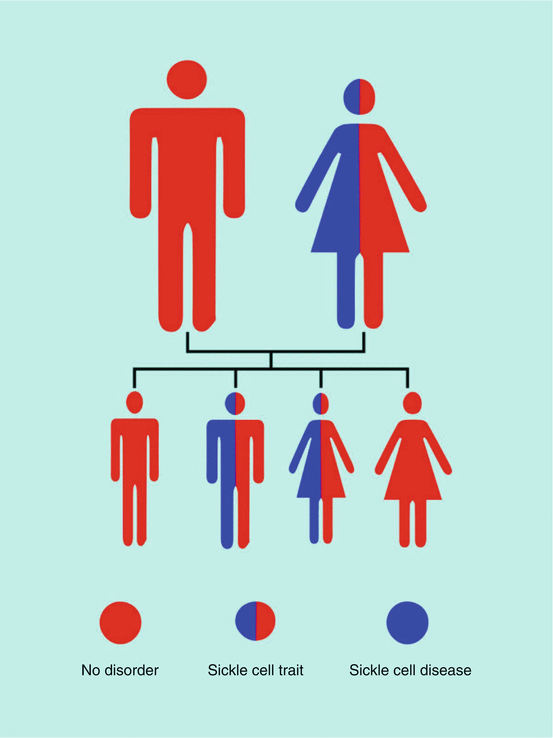
If one parent has sickle cell trait (one normal gene and one sickle cell gene) and the other parent has two normal hemoglobin genes (Fig. 2.14):
Related posts:
 Recent Advances in the Treatment of Sickle Cell Anemia
Hydroxyurea Treatment for Sickle Cell Anemia
Recent Advances in the Treatment of Sickle Cell Anemia
Hydroxyurea Treatment for Sickle Cell Anemia
 Perioperative Management of Patients with Sickle Cell Anemia
Perioperative Management of Patients with Sickle Cell Anemia
 Cardiovascular Complications of Sickle Cell Anemia
Cardiovascular Complications of Sickle Cell Anemia
 The Acute Chest Syndrome in Sickle Cell Anemia
The Acute Chest Syndrome in Sickle Cell Anemia
 Hepatobiliary Complications of Sickle Cell Anemia
Hepatobiliary Complications of Sickle Cell Anemia

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


