The identification of dermatologic abnormalities and their association with internal malignancies often require thorough observation from clinicians. A consultation with a dermatologist may be helpful to identify specific dermatologic abnormalities. In some cases, biopsy and pathology may be necessary for a diagnosis.
Genetic counseling for hereditary skin diseases is similar to the process for other cancer predisposition syndromes. The genetic counseling process generally includes a detailed family and medical history, risk assessment, discussion of benefits, and limitations of available genetic testing, including possible test results, discussion of medical management, and implications for family members.19 Dermatologic evaluation and review of pathology records pertaining to the cutaneous findings may provide clarification on specific dermatologic observations. Consultation with a dermatologist and/or other specialist who is knowledgeable about hereditary syndromes is often essential to a clinical evaluation. When possible, reviewing the medical records of family members is also helpful to confirm dermatologic diagnoses, as reports of some skin findings in family members may contain some inaccuracies.20
HEREDITARY SKIN CANCER AND THE NEUROFIBROMATOSES
In addition to a few known single-gene disorders associated with skin cancers, confounding environmental factors, including solar ultraviolet radiation, as well as other genetic factors also are known to be associated with a varying degree of skin cancer risk. Separately, other hereditary tumor and cancer predisposition syndromes, such as NF1 and NF2, contain benign cutaneous features as common and sometimes predominant findings. General characteristics of a hereditary cancer predisposition syndrome include multiple tumors or cutaneous features in one individual, multiple affected family members, and individuals or families with related tumors, cancers, or unique physical characteristics. In some cases, young age at onset may also suggest a higher likelihood of a hereditary syndrome.
Hereditary Melanoma
Approximately 10% of melanoma cases are attributed to hereditary predisposition. Hereditary melanoma has been associated with mutations in two genes, cyclin-dependent kinase inhibitor 2A (CDKN2A) and cyclin-dependent kinase 4 (CDK4). Mutations in CDK4 are rare and have been identified in only a few hereditary melanoma families.21 Of families with hereditary melanoma, defined as three or more diagnoses of melanoma in one family, approximately 20% to 40% will have a detectable mutation in CDKN2A.22
CDKN2A and CDK4 both function as tumor suppressors. CDKN2A encodes two transcripts: p16 and p14ARF through alternate reading frames. The majority of CDKN2A mutation-carrying families have been found to have mutations that affect the p16 protein. Mutations affecting the function of p14ARF are reportedly rare in cutaneous melanoma families.23
Phenotype
Hereditary melanoma has also been referred to as familial atypical mole melanoma syndrome.24 Although the presence of atypical moles has been associated with an increased risk for melanoma, it has not been identified as a strong predictor of CDKN2A mutation status.25,26
The penetrance of CDKN2A mutations has been observed to be dependent on geography. This is likely due to varying environmental and other genetic factors across geographic regions. A study of CDKN2A carriers selected based on positive personal and family history of melanoma observed the melanoma risk for CDKN2A mutation carriers to be 58% in Europe, 76% in the United States, and 91% in Australia.27 In a population-based study of patients with melanoma, the penetrance of CDNK2A mutations was observed to be lower (28% risk for melanoma by the age of 80 years).28 Variants in the melanocortin 1 receptor (MC1R) gene have been associated with increased CDKN2A penetrance.29 The prevalence of MC1R has been observed to differ with ethnic background and is one example of a genetic factor influencing melanoma risk that varies by geographical region.30
In addition to melanoma, other cancers have also been observed in increased frequency in CDKN2A mutation carriers. Most notably, an increased risk for pancreatic cancer has been reported in some CDKN2A mutation–carrying families.31 Less commonly, an increased risk for other cancers, including neural system tumors, nonmelanoma skin cancers, uveal melanoma, and head and neck cancers, has also been reported in individuals with CDKN2A mutations.31,32
In the United States, which is an area of moderate to high melanoma incidence, genetic counseling for hereditary melanoma has been generally recommended in families in which (1) three or more relatives are affected with melanoma, (2) one individual has three or more primary melanomas, or (3) both pancreatic cancer and melanoma are present in one family (Table 95.2).15 Early age at onset in the absence of a family history of melanoma is not highly suggestive of a CDKN2A mutation.33,34

Genetic Testing
Clinical testing for CDKN2A and CDK4 is available in the United States at several commercial laboratories. However, some of the laboratories offering hereditary melanoma testing perform analysis of only CDKN2A, given the relatively low-frequency CDK4 mutations reported.
The utility of genetic testing for CDKN2A mutations remains a source of debate. This is partly due to the relatively low frequency of CDKN2A mutations in families with melanoma. In addition, many individuals with a personal and/or family history of melanoma are under close surveillance and aware of risk-reduction recommendations; therefore, genetic test results would not alter clinical management.25 Also, the role of pancreatic cancer surveillance in CDKN2A carriers remains under investigation. Some studies have suggested that knowledge of CDKN2A mutation status improves short-term compliance to risk-reducing behaviors.35,36 However, information regarding the long-term impact of CDKN2A testing is limited at this time. The possible genetic test results for an individual undergoing CDKN2A genetic testing are shown in Table 95.3.
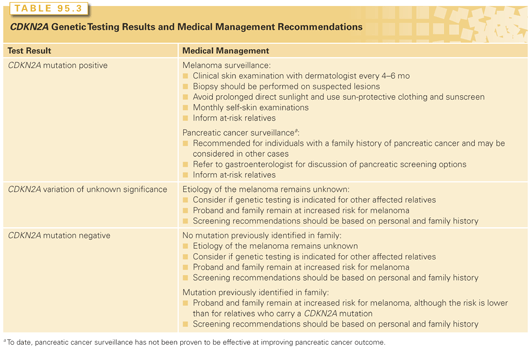
Individuals with a CDKN2A mutation have a 50% chance of passing the mutation on to their children.
Medical Management
CDKN2A mutation carriers, or individuals at 50% risk to be a carrier, should be monitored carefully for melanoma through clinical and self-examinations (see Table 95.3). In addition, CDKN2A carriers are recommended to avoid prolonged direct sunlight and utilize sun-protective clothing and sunscreen.25,37
Individuals who test negative for a familial CDKN2A mutation may also have an increased risk for melanoma. However, this risk has been observed to be lower than the melanoma risk for CDKN2A mutation carriers.28
As noted in Table 95.3, CDKN2A mutation carriers, especially those with a family history of pancreatic cancer, are candidates for pancreatic cancer surveillance and should discuss the risks, benefits, and limitations of screening with a gastroenterology specialist.38 However, to date, the effectiveness of pancreatic surveillance remains under investigation.39
Basal Cell Nevus Syndrome
BCNS, also known as Gorlin syndrome or nevoid basal cell carcinoma syndrome, is an autosomal dominant syndrome associated with cutaneous findings, including basal cell carcinoma, as well as skeletal system, nervous system, and ocular abnormalities.40 Although BCNS has complete penetrance, the expression is variable.41
BCNS is thought to be relatively uncommon, and the incidence of BCNS has been estimated to be 1:30,827 to 1:57,000.42 The variable expression may cause difficulty in diagnosing BCNS.
BCNS has been associated with mutations in the patched gene 1 (PTCH1) gene. PTCH1 functions as a tumor suppressor in the sonic hedgehog (Shh) pathway, which is also involved in embryonic development.43 Chromosomal abnormalities of 9q22.3 region, which includes PTCH1, have been reported in a few individuals with features of BCNS as well as other features, including short stature, developmental delay, and seizures.44 Rarely, mutations in other genes, including SUFU and PTCH2, have also been reported in individuals with features of BCNS.45,46
Phenotype
The phenotype of BCNS is variable, and some characteristics are present at different life stages. Therefore, it is important to obtain a complete medical history, including physical examination and dermatologic, cardiac, and gynecologic examinations as well as radiologic studies to confirm a diagnosis of BCNS.
The clinical manifestations of BCNS include the following.
Skin
Basal Cell Carcinoma. Approximately 50% to 75% of individuals with BCNS will develop basal cell carcinomas.47 Typically, basal cell carcinomas develop in the late teens through the 30s, but some published reports have indicated the detection of basal cell carcinomas in early childhood in individuals with BCNS. The presence of basal cell carcinomas is also dependent on other factors, including skin type and radiation exposure, including sun exposure.40,41
Noncancerous Cutaneous Features. The majority of individuals with BCNS will have multiple nevi present by adulthood.40 In addition, BCNS is associated with an increased prevalence of facial milia, dermoid cysts, and skin tags. Palmar and plantar pits are also a common feature of BCNS and usually are evident by early adulthood.40
Skeletal
Skeletal abnormalities, including rib and spinal abnormalities, are reported with increased frequency in BCNS. The majority of individuals with BCNS are reported to have macrocephaly.48
Central Nervous System
Ectopic Calcification. Ectopic calcification, particularly of the falx celebri, has been reported as a common finding in individuals with BCNS.48
Brain Tumor. Although other types of brain tumors have been reported in individuals with BCNS, medulloblastoma, typically desmoplastic type, is the most common.49 Approximately 5% of individuals with BCNS are diagnosed with medulloblastoma, usually around 2 years of age.
Other Features
Jaw Keratocysts. Approximately 75% of affected individuals with BCNS develop multiple jaw keratocysts.50
Characteristic Facial Features. Facial features characteristic of BCNS, including macrocephaly, bossing of the forehead, coarse facial features, and facial milia, have been observed in approximately 60% of BCNS cases.14
In addition to these features, congenital malformations such as cleft lip/palate, polydactyly, and eye anomalies have also been reported as features of BCNS.40
Additional associated tumors including cardiac and ovarian fibromas have also been reported to occur with increased frequency in BCNS.51,52
Diagnosis and Genetic Testing
A diagnosis of BCNS was initially based on clinical criteria; however, the availability of molecular testing has identified mutations in individuals with a more variable phenotype. The First International Colloquium on BCNS concluded that the clinical criteria should be used to consider a suspected diagnosis of BCNS rather than as diagnostic criteria.53 The colloquium recommends that a suspected diagnosis of BCNS be considered in individuals with an identified PTCH1 mutation and one major clinical criterion, individuals who express two major criteria, and individuals with one major and two minor criteria (Table 95.4).

Genetic testing for the PTCH1 gene is clinically available. Approximately 50% to 85% of individuals with clinical features of BCNS will have a detectable mutation in the PTCH1 gene through gene sequencing analysis. Deletions and duplications of the PTCH1 gene have also been reported.54
Approximately 20% to 30% of individuals with BCNS are de novo, meaning that neither parent carries the associated gene mutation.14 Individuals affected with BCNS have a 50% chance of having an affected child. In cases where a mutation has been identified, testing is an option for at-risk family members. In addition, both preconception genetic diagnosis and prenatal testing are available for known PTCH1 mutations.
Medical Management
Because of the many variable symptoms of BCNS, individuals with BCNS should be referred to an appropriate specialist depending on the symptoms.
Basal Cell Carcinoma. Early diagnosis is important for management and to limit cosmetic damage. Surgery, oral retinoids, topical therapies, and photodynamic therapy have all been utilized with varying degrees of success for individuals with BCNS.47
Medulloblastoma. Consideration of developmental assessment and physical examination every 6 months is an option for children during infancy and early childhood. Imaging for medulloblastoma surveillance is not currently recommended.14
Jaw Keratocysts. Clinical examinations and imaging are recommended for individuals with BCNS, starting during childhood. These tumors may sometimes be detected during routine dental examinations.55
Ovarian and Cardiac Fibromas. Affected individuals with cardiac fibromas should be referred to a cardiologist. Ovarian fibromas also warrant a specialty referral and may require surgery, ideally with the aim of preserving fertility.56
Radiation Exposure. Given the known increased risk for basal cell carcinoma, it is recommended that individuals with BCNS avoid sun exposure. In addition, it is recommended that other radiation exposure also be avoided if possible, including radiation as treatment for medulloblastoma.49
Neurofibromatosis Type 1
NF1 is one of the most common genetic disorders, affecting an estimated 1:2,500 to 1:3,000 individuals at birth.7 Formerly known as von Recklinghausen disease or peripheral neurofibromatosis, manifestations of the disease affect multiple areas of the body, including, but not limited to, the central and peripheral nervous systems, skin, eyes, skeleton, gastrointestinal system, and the cardiovascular system. Historically, observations of patients with NF1 date back to the 13th century, but the disorder was first formally described in 1882 by Friedrich von Recklinghausen.7,57,58
NF1 is a completely penetrant autosomal dominant condition with widely variable expression, both within and between families.59 No ethnic, racial, or sex predilection has been observed.57 NF1 is caused by mutations in the NF1 gene on 17q11.2. The protein product of NF1 is neurofibromin, a GTPase-activating protein that is expressed across many tissue types and in particularly high levels within neurocutaneous tissue. It acts as a negative regulator of intracellular Ras signaling pathways involved in cell growth and proliferation.7,60,61 More recently, NF1 has also been linked to the development of skeletal muscle.62
Phenotype
In 1987, the National Institutes of Health developed clinical diagnostic criteria for NF1 (Table 95.5
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


