Genetic Regulation of Islet Function
Michael S. German
Despite their small size and apparent simplicity, pancreatic islets of Langerhans are remarkably sophisticated micro-organs. The four cell types that comprise each islet have in common basic endocrine-cell characteristics and are thought to derive from a common precursor during development. In the mature pancreas, however, each cell type functions quite differently, responding to different signals and producing different secreted products. These marked differences result from the differential expression of a unique set of genes. The mechanisms that control differential gene expression determine the functional characteristics of the islet cells (1,2,3).
The genes differentially expressed in the islet cells include the peptide hormone genes such as those encoding preproinsulin and proglucagon but also include a variety of genes involved at all levels of cell function, such as glucokinase, glutamic acid decarboxylase, the prohormone convertases, cell surface receptors, and cell adhesion molecules. Clearly, a complex regulatory network is needed to ensure that the complete set of genes is expressed at appropriate levels. This regulatory network in turn must quickly mature as each islet cell type develops from a common cluster of pluripotent progenitor cells. Understanding the genetic control of islet function requires an understanding of both how this network emerges during the differentiation of the islet cells and how it functions in the mature islet cell.
DIFFERENTIATION OF DISTINCT ISLET CELL TYPES DURING DEVELOPMENT
As described in the preceding chapter, the islet cells of the pancreas, along with the exocrine and duct cells, differentiate from a common pool of undifferentiated progenitor cells that form the initial pancreatic buds. This process of differentiation results from serial changes in gene expression as uncommitted precursor cells progress to mature, postmitotic, terminally differentiated cells (1,2,3,4,5,6).
Several models for the lineage relationships of pancreatic endocrine cells have been proposed, generally based on the order of appearance of cell types during fetal development. For example, because the earliest endocrine cells in the fetal pancreas express glucagon, it has been proposed that α-cells function as precursors for the other islet cell types that appear later (7,8). In the absence of labeling studies, however, order of appearance cannot establish a lineage relationship. In fact, a preponderance of recent evidence strongly suggests that α-cells are not precursors for β-cells. First, the early glucagon-expressing cells are postmitotic and thus cannot provide an explanation for the later appearance of much larger numbers of β-cells without cell replication (9,10,11). Furthermore, during the peak of formation of new β-cells (around embryonic day [E] 15 in the fetal mouse), no cells representing a transition between α- and β-cells with coexpression of glucagon and insulin can be detected (7,8). Finally, experiments with transgenic mice with toxic genes or cre recombinase labeling demonstrate that β-cells do not develop from a precursor in which the glucagon promoter is active (12,13,14). Together, this evidence supports a model in which islet cell type is determined before the expression of the hormone genes.
Insulin-producing cells may develop via more than one pathway. The earliest insulin-producing cells in the mouse fetus, those appearing before E13, do not express the normal set of β-cell genes, because they lack GLUT2, express low levels of insulin, and may coexpress glucagon. These cells do not replicate, and their numbers are small; thus, it is unlikely that they contribute significantly to the population of β-cells in the mature islet. It still remains to be determined whether all of the β-cells present in the mature pancreas differentiated via a common pathway; but because they express the same restricted set of genes, the mechanisms that control gene expression are likely to be the same.
Despite their obvious differences, the four different islet cell types share similar developmental pathways and related functions, and these connections are reflected in similarities in gene-expression mechanisms and in an overlapping set of expressed genes, some of which are also expressed in pancreatic ductal or exocrine cells. Because of its central role in energy metabolism, along with its robust expression and tight restriction to β-cells, the insulin gene has emerged as a paradigm of cell type-restricted expression in the endocrine pancreas.
THE INSULIN-GENE PARADIGM
Humans have a single insulin gene, located on chromosome 2, at 2p21 between the tyrosine hydroxylase (THI) gene and the insulin-like growth factor-2 (IGF-2) gene (Fig. 4.1) (15). The gene contains two introns, one interrupting the 5′ untranslated sequence and the second interrupting the sequence encoding the C-peptide. This general structure is conserved, with few exceptions, among the other mammalian insulin genes, as well as among the related IGF genes (16).
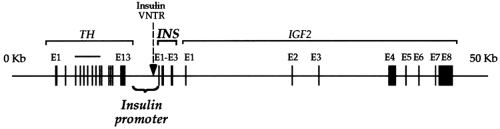 Figure 4.1. The human insulin gene (INS) and surrounding loci on chromosome 11. IGF2, insulin-like growth factor-2; TH, tyrosine hydroxylase. |
The sequences upstream of the transcription start site comprise the insulin-gene promoter (17). The promoter directs RNA polymerase II to the correct transcription start site, restricts insulin-gene transcription to the β-cell, and responds to metabolic, hormonal, and neural signals by modulating the rate at which insulin-gene transcription is initiated. These functions of the insulin-gene promoter are relatively independent of chromosomal context: When removed from the remainder of the insulin gene, the promoter can similarly regulate any gene inserted downstream.
The use of such chimeric genes, with the isolated insulin promoter driving the expression of an easily detectable marker gene such as chloramphenicol acetyl transferase, SV40 large T antigen, or firefly luciferase (18,19,20,21,22,23,24), provides a simple method of testing the importance of specific sequences within the promoter. When introduced into insulin-producing cells in culture or into transgenic mice, these chimeric genes demonstrate that a relatively small fragment of the insulin promoter, the proximal 250 to 400 base pairs (bp), provides the same regulation of transcription observed with the intact gene, although larger promoters increase the fidelity and the relative level of transcription in transgenic animals (25).
Careful mutational analyses within the proximal promoter have further delineated the specific sequences that contribute to the full activity of the promoter (22,23,24,26,27). Figure 4.2 outlines the general architecture, including functionally important sequences in the human and rat insulin-gene promoters. In some regards, the insulin-gene promoter is structurally similar to many cellular gene promoters and contains promoter elements that can function in all cell types. These include a consensus TATAA box, the binding site for the TFIID complex, located 30 bp upstream from the transcription start site, a CCAAT box, and one or more cyclic adenosine monophosphate regulatory elements (27,28,29,30).
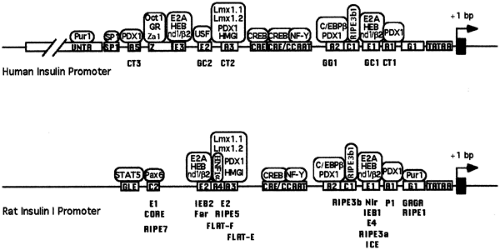 Figure 4.2. The human insulin gene and rat insulin I gene promoters with known sequence elements and binding factors. The boxes represent characterized sequence elements. The binding proteins are circled above the promoter. Older names for the sequence elements are listed below the promoter. GR, glucocorticoid receptor; nd1/β2, neuroD1/BETA2. |
Other portions of the promoter, however, are more unique and give the promoter the specificity that limits its activity to the β-cells. As shown in Figure 4.2, a number of sequence elements along the proximal promoter contribute to this specific activity, but comparison of the available mammalian insulin-gene promoter sequences reveals certain common themes. Most prominently, all of the mammalian insulin-gene promoters contain
juxtaposed E and A sequence elements. The E elements contain the core sequence motif CCANCTG, and the A elements contain A/T-rich sequences with one or more copies of the sequence TAAT.
juxtaposed E and A sequence elements. The E elements contain the core sequence motif CCANCTG, and the A elements contain A/T-rich sequences with one or more copies of the sequence TAAT.
Function of the human and rat insulin promoters depends on the presence of at least one A element and one E element. Mutation of either of the two E elements in the rat insulin I gene results in a 90% loss of promoter activity in insulinoma cells, while mutation of both elements ablates all activity (26). The more proximal E element, E1, is absolutely conserved in all known mammalian insulin genes, and its importance has also been demonstrated in the rat insulin II promoter and the human insulin promoter (23,26,31,32). The A3 and A1 elements are both highly conserved in mammalian insulin genes, and the A3 element has been found to play an important role in both rat insulin-gene promoters and the human insulin promoter (23,26,31,32).
The Basic Helix-Loop-Helix Transcription Factors: NeuroD1
The sequences of the insulin E elements are a subset of the common CANNTG E box elements that play important roles in many cell type-specific promoters, including the promoters for the immunoglobulin genes and muscle-specific genes (33). E boxes are binding sites for proteins of the basic helix-loop-helix (bHLH) family. The bHLH proteins form dimers through their helix-loop-helix domains, and these dimers contact and bind to DNA with the basic domains. Because heterodimerization is permitted, and in most cases favored, a large variety of complexes are possible. Cell type-specific complexes generally result from heterodimerization of one of a limited number of ubiquitously expressed bHLH proteins (class A) with one of a large set of cell type-restricted bHLH proteins (class B).
β-Cells contain several bHLH complexes capable of binding to the insulin E boxes. Nuclear extracts from insulinoma cells demonstrate that the predominant complexes are formed from dimerization of a ubiquitous bHLH protein (the E12 or E47 products of the E2A gene or HEB) with the neuroendocrine bHLH protein neuroD1/BETA2 (24,34,35,36,37,38,39). NeuroD1 is expressed in all of the islet cells, in the endocrine cells of the gut, and in the central nervous system. The carboxyl-terminus of neuroD1 contains a potent transcriptional activation domain (40,41) (Fig. 4.3), and ectopic expression of neuroD1 can activate the insulin promoter in non-β-cells (38).
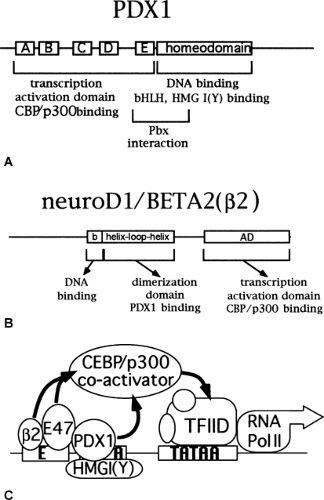 Figure 4.3. The insulin promoter transcription complex. A: The functional domains of the PDX1 protein are outlined. The regions labeled A-E represent conserved domains within the activation domain (90) B: The functional domains of the neuroD1/Beta2 protein are outlined. C: Interactions are shown among PDX1, the bHLH proteins, HMGI(Y), and p300 that result in transcriptional activation of the E-A mini-enhancer. β2, neuroD1/BETA2. |
Mice homozygous for a targeted disruption of the gene encoding neuroD1 have a marked decrease in islet cells due to premature apoptosis and die of diabetes shortly after birth (42,43). In addition, absence of neuroD1 results in losses of enteroendocrine cells and in defects in several aspects of neural development (42,43,44,45,46,47,48,49). Interestingly, insulin is still produced by the remaining β-cells in the pancreas, suggesting either that neuroD1 function is not required for insulin-gene transcription or that other bHLH proteins may be able to substitute for neuroD1 on the insulin promoter (42).
Homeodomain Transcription Factors: PDX1
The A elements function as binding sites for homeodomain proteins, a broad family of transcription factors. Homeodomain proteins have been found in all eukaryotic organisms and are characterized by a conserved stretch of 61 amino acids called the homeodomain, which forms a helix-turn-helix structure that binds to DNA. Homeodomain proteins play critical roles in cell-type determination and differentiation during development, as well as in the regulation of gene expression in the mature cells.
In extracts from β-cell nuclei, the most abundant protein binding to the A elements is the homeodomain protein PDX1 (also called IPF1, IDX1, STF1, IUF1, and GSF1) (50,51,52,53,54,55,56). PDX1 belongs to the parahox family of homeobox genes and is expressed in the antral stomach, duodenum, and pancreas (50,57). Early in development PDX1 is expressed broadly in the pancreatic bud, but after E13 in the mouse, it becomes restricted, with high levels of expression in β-cells and some δ-cells and lower levels of expression in acinar cells and duct cells. PDX1 can bind to all of the A elements in the rat and human insulin promoters and can activate portions of the insulin promoter in cell lines (50,51,52). Furthermore, forced expression of PDX1 has been reported to activate the endogenous insulin gene in some non-β-cells (58,59).
Mice homozygous for a targeted disruption of the gene encoding PDX1 have a profound defect in pancreatic growth, with essentially no development of the pancreas beyond initial bud formation, and die shortly after birth (60,61,62). A few hormone-producing cells, including insulin-producing cells, however, can be detected in the remaining dorsal bud (61). If instead
the pdx1 gene is inactivated specifically in β-cells after the pancreas has formed, by using a cre-lox recombination system with the insulin promoter driving the cre recombinase, the differentiated phenotype of the β-cells is partially lost (63). In these animals with a β-cell-specific disruption of the pdx1 gene, the β-cell mass decreases; and the remaining β-cells coexpress glucagon and have reduced GLUT2 and reduced glucose-stimulated insulin secretion. As in the β-cells lacking neuroD1, however, the β-cells lacking PDX1 continue to produce insulin.
the pdx1 gene is inactivated specifically in β-cells after the pancreas has formed, by using a cre-lox recombination system with the insulin promoter driving the cre recombinase, the differentiated phenotype of the β-cells is partially lost (63). In these animals with a β-cell-specific disruption of the pdx1 gene, the β-cell mass decreases; and the remaining β-cells coexpress glucagon and have reduced GLUT2 and reduced glucose-stimulated insulin secretion. As in the β-cells lacking neuroD1, however, the β-cells lacking PDX1 continue to produce insulin.
The persistence of insulin-gene transcription in the absence of PDX1 suggests the presence of additional homeodomain proteins in β-cells that can drive insulin-gene expression in vivo. Although it is clearly the most abundant nuclear protein capable of binding the A elements in β-cells, PDX1 is not the only homeodomain transcription factor expressed in β-cells nor the only homeodomain protein in β-cells capable of binding and activating transcription through the A elements (54,64,65,66). Two β-cell homeodomain proteins, Cdx2/3 and lmx1A, have been shown to bind to and activate through the A3 and A4 sites in the rat insulin I promoter (65). In addition, the POU-homeodomain protein HNF1a is also expressed in β-cells, and it binds to and activates the A4 site in the rat insulin I promoter, although the A4 site is not conserved in insulin genes from other species (66). Finally, β-cells express a diverse set of additional homeodomain transcription factors; some of these regulate insulin-gene transcription, but all play some part in β-cell gene expression (54).
Paired-Homeodomain Factors: Pax6
The E and A elements and the protein complexes that bind to them are essential to the function of the insulin promoter but cannot explain all of the activity of the promoter. A G/C rich sequence, C2, in the distal promoter contributes significantly to the activity of the rat insulin promoters both in insulinoma cells and primary-cultured β-cells (26,27,31). Because the C2 element and related elements in the glucagon and somatostatin gene promoters all function in an islet cell-specific manner and bind to an islet-specific complex, this common islet enhancer element has been termed PISCES (pancreatic islet cell-specific enhancer sequence) (67,68,69,70,71,72).
C2 and the other PISCES elements closely match the consensus binding sequence for the paired-homeodomain transcription factor Pax6, and Pax6 antiserum recognizes the islet-specific complex that binds to all three PISCES elements (73). Pax6 contains a potent transcriptional activation domain and three DNA-binding domains, a paired region composed of the pai and red domains and a homeodomain (74,75,76). Pax6 protein first appears during development in the nuclei of a few scattered cells in the gut endoderm where the dorsal bud and the first glucagon-producing cells will appear a few hours later (73). From that point on, it is expressed in the nuclei of all the endocrine cells in the pancreas, as well as in a subset of neuroendocrine cells in other tissues.
Mice carrying mutations in the pax6 gene were previously identified and named small eye because of a defect in eye development in the heterozygous mice. Mice homozygous for the small-eye mutations or a targeted interruption of the pax6 gene die at birth with multiple craniofacial and neurologic defects. In addition, the homozygous mutant animals have profound defects in islet development, with decreased numbers of all islet cell types (73,77). The remaining β-cells also have a significant decrease in the amount of insulin mRNA and protein per cell. These mice demonstrate that Pax6 is important but not absolutely essential for expression of the insulin gene in mice. The importance of Pax6 in human insulin-gene expression is less certain, however, because the C2 element is not conserved in the human insulin-gene promoter.
Unidentified Factors: RIPE3b1
Immediately upstream of the E1 element lies the C1 element, a cytosine-rich sequence that is conserved in all of the mammalian insulin promoters. The C1 element is critically important for the function of both the human insulin and rat insulin II promoters (31,32,35,78,79,80). A protein complex found in β-cell nuclei, the RIPE3b1 complex binds specifically to the C1 element and has not been detected in other cell-types, including α-cells (35,81). Although it is known that the RIPE3b1 complex is composed of several proteins of similar molecular mass and that binding to the C1 element requires its phosphorylation, the protein has not been identified (82,83,84).
Unidentified Factors: Za1
Upstream of the A3 element in the human insulin promoter lies the negative regulatory element (NRE), which strongly inhibits insulin promoter function in β-cell tumor lines (32,85,86). The NRE also inhibits transcription when isolated from the insulin promoter and linked to a heterologous promoter in either β-cell or non-β cell tumor lines (32,85). It has been proposed that the NRE may modulate insulin promoter activity and help restrict insulin expression to the β-cell (85).
The NRE may not always function, however, simply as a silencer of transcription. Walker et al. (18), for example, found that removal of the NRE from the human insulin promoter had no significant effect on the promoter, and Clark et al. (85) found that isolated sequences within the NRE could activate transcription weakly. It is in primary cultured islets, however, that the NRE reveals its most remarkable activity. Removal of the NRE from the human insulin promoter causes a marked loss of activity in cultured fetal rat islet cells (80). Placed upstream of a minimal insulin promoter or a heterologous promoter, the NRE functions as a potent activator of transcription in both fetal and adult islets, whereas in the same constructs it strongly represses transcription in tumor cell lines from β-cells or non-β cells and in primary-cultured fibroblasts (86). Because it appears that the primary function of this region in normal β-cells in vivo is to activate transcription, this region has been renamed the Z element. Although a Z element-binding complex that correlates with the positive activity of the Z element can be detected in nuclear extracts from primary cultured islets (86), the proteins in this complex have not been identified.
The Insulin Promoter Transcription Complex
The linear view of the insulin promoter as a series of binding sites and binding proteins, as shown in Figure 4.2, fails to illustrate the complexity and dynamic character of the functioning promoter in vivo. Missing from this view is the added dimension provided by the interactions among the transcription factors, DNA structural proteins, co-activators and repressors, intracellular signaling molecules, and the RNA polymerase complex. This view also ignores the role of upstream factors that regulate the expression and function of these proteins. The promoter should be regarded as a large and fluid complex formed by interactions among many different proteins and the chromosome in dynamic equilibrium with forces acting from within and outside the cell.
In this perspective, the bipartite E-A combination represents the minimal functional component of the larger complex and demonstrates the underlying concept of cooperativity. Neither
the E elements nor the A elements by themselves have any significant transcriptional activity in β-cells; but together they can dramatically boost the activity of a linked promoter in a β-cell-specific fashion (87,88). The synergy between these elements results from interactions among the proteins that bind to them and non-DNA-binding transcriptional co-activators.
the E elements nor the A elements by themselves have any significant transcriptional activity in β-cells; but together they can dramatically boost the activity of a linked promoter in a β-cell-specific fashion (87,88). The synergy between these elements results from interactions among the proteins that bind to them and non-DNA-binding transcriptional co-activators.
This cooperativity can be demonstrated when PDX1 is coexpressed along with either E47 or E47 plus neuroD1 in non-β-cells: activation of the E-A mini-enhancer depends on the combination of PDX1 bound to the A element and a bHLH dimer bound to the E element (89,90). In addition to both the E and A binding sites, cooperativity between PDX1 and the bHLH dimer requires the DNA-binding domains of all the proteins and the transcriptional activation domains of both PDX1 and at least one of the bHLH proteins (89,90,91,92,93).
Cooperativity results from interactions among the proteins at several levels (Fig. 4.3). First, the proteins cooperate at the level of DNA binding. Physical interaction among the DNA binding domains, the homeodomain of PDX1, and the bHLH domains of E47 and neuroD1/BETA2, stabilizes the complex of all three proteins on DNA. Although DNA binding affinity is increased, with purified proteins in vitro the relative increase in binding affinity is small (92). In vivo, however, the nuclear environment is very different, with differences in local concentrations of factors and the presence of many other proteins, all of which change the energetic equilibrium of DNA binding. In addition, chromatin structure is quite different from the purified oligonucleotides used to test DNA binding in vitro. Chromosome structure is controlled in vivo by the binding of histones and of small, abundant DNA-binding proteins, the high-mobility group (HMG) proteins.
The presence of these structural DNA-binding proteins changes the energetic cost of DNA-binding by the transcription factors (for a review, see reference 94). For example, in the presence of HMG I(Y), a small nonhistone DNA-binding protein that can bind to the A3/4 region of the insulin promoter, co-binding of PDX1 and the bHLH dimer is greatly favored in vitro. As a result, HMG I(Y) increases PDX-bHLH transcriptional synergy in vivo. In insect cells, where HMG I proteins are expressed at very low levels, PDX1 and the bHLH proteins cannot cooperatively activate the E-A mini-enhancer without the addition of HMG I(Y) (92).
At the next level of organization, the insulin promoter activation complex requires non-DNA-binding proteins that form a bridge between the proteins binding at the E and A sites and the basal transcriptional machinery. Just as interactions within the PDX1/bHLH/HMG I(Y) complex stabilize DNA binding and increase the occupancy of the E-A sites on the insulin promoter, multiple protein-protein contacts stabilize the interaction with the basal transcription machinery and ensure efficient transcription initiation. The grouping of transcription factors on adjacent DNA sites, as in the E-A mini-enhancers, creates clusters of protein interaction sites that cooperatively recruit or stabilize binding of the RNA polymerase II transcription initiation complex (95). Non-DNA-binding co-activators promote this process by linking the DNA-bound transcription factors with TFIID complex and the basal transcription machinery. The additional interactions provided by the co-activators provide critical stability and play a linchpin role in the formation and the function of the insulin promoter transcription-activation complex (Fig. 4.3). In support of this model, the E2A proteins, neuroD1, and PDX1 have all been shown to interact with the ubiquitous p300 co-activator (40,41,47,96,97).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


