In 1860, Dr. Broca, a French surgeon, reported on the pedigree of his wife’s family because of her early-onset breast cancer and the striking family history of breast cancer.1 At around the same time, in 1865, Dr. Gregory Mendel was looking at the genetics of garden peas; his work would lay the foundation for the field of genetics. In the last 150 years, the identification and recognition of the molecular genetics component of disease has advanced, and the area of breast cancer has been no exception. Molecular genetic testing is redefining familial cancer and will continue to do so for many years. This chapter will review the clinical features, causative genes, and medical management options for hereditary breast cancer syndromes.
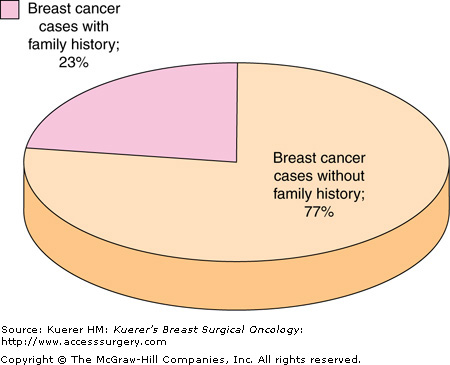
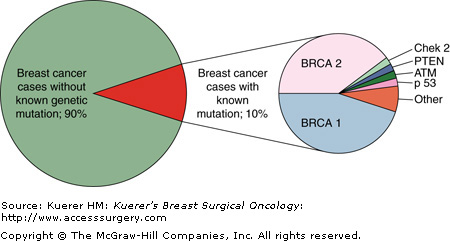
It has been estimated that 23% of all breast cancer patients have a family history of the disease, indicating that one or more first-degree relatives have been diagnosed with breast cancer in the family (Fig. 8-1).2 In the general population, only 2% of all women have a family history suggestive of the inheritance of an autosomal dominant, inherited high-penetrance gene for breast cancer.2 When you look specifically at breast cancer patients, only 5% to 10% of all breast cancer cases demonstrate a clear pattern of dominant inheritance.4 There are several highly penetrant, dominantly inherited genes associated with breast cancer predisposition syndromes, including the BRCA1, BRCA2, TP53, and PTEN genes (Fig. 8-2). Although these cancer predisposition syndromes are rare, the clinician needs to be aware of these hereditary cancer syndromes that predispose to the carcinoma of the breast, and to recognize the diversity of these unique syndromes. However, most cases of familial breast cancer are not attributed to these single-gene disorders. It is believed that may are due to unidentified, low-penetrant genetic mutations and/or variants in the genome, in combination with environmental factors. The identification of these low-penetrant alleles is the next frontier for cancer genetics.
The American Society of Clinical Oncology recommends that genetic testing be offered when three criteria are met: (1) the individual has a personal or family history suggestive of a cancer genetic susceptibility condition, (2) the test can be adequately interpreted, and (3) the results will aid in the diagnosis or influence the medical or surgical management of the patient or family members.5 These recommendations begin with the identification of these high-risk patients. A comprehensive review of family history is key to identifying patients at increased risk for hereditary cancer. In obtaining a 3-generation family history, it is important not only to elicit the family history of breast cancer, but to also ask about other cancers, including ovarian, thyroid, sarcomas, adrenocortical carcinoma, endometrial, pancreatic, brain tumors, and dermatologic manifestations. It is critical to obtain a family history on both the maternal and paternal side of the family. Also, one must take into consideration reduced penetrance, limited family medical information, and small family size. Besides recording the types of cancer in the family, the National Comprehensive Cancer Network (NCCN) guidelines recommend that the clinician determine not only the types of cancer in the family, but the age of diagnosis, documentation of the diagnosis from medical records (particularly pathology reports of primary cancers), a history of chemoprevention and/or risk-reducing surgery, carcinogen exposure, reproductive history, hormone use, and previous breast biopsies.6 The family and patient’s medical history, in conjunction with computerized risk assessment models, can quantify the patient’s risk for a genetic predisposition to breast cancer.7-9
Several organizations have proposed criteria for when a patient should be referred for genetic counseling and testing regarding hereditary breast and ovarian cancer. Although the criteria may vary slightly, they all recognize similar features: early-onset breast cancer (premenopausal or age 50 or less), ovarian cancer (with a family history of breast or ovarian cancer), individuals with two or more primary breast cancers, breast and ovarian cancer in the same individual, male breast cancer, and Ashkenazi Jewish ancestry. In 2006, the National Comprehensive Cancer Network published practice guidelines for Genetic/Familial High-Risk Assessment: Breast and Ovarian Cancer.6
Cancer genetic counseling involves education regarding the characteristics of hereditary cancer, the patient’s personal risk of developing cancer, and the risk that the patient carries a mutation in a cancer predisposition gene. The key components to the genetic counseling process have been outlined by the National Society of Genetic Counselors (NSGC).10,11 Genetic counselors discuss the benefits, limitations, and risks of genetic testing, as well as options for cancer prevention and management. Genetic counseling should also include a discussion of testing sensitivity and specificity, inconclusive genetic results, and variants of uncertain clinical significance. The goal of the process is to communicate the complex issues involved in genetic testing in a language that the patient understands and in order for the patient to make informed decisions about genetic testing and his or her health care. Attention to the psychosocial issues is critical to this process in order to initiate health care behavior changes. Genetic counseling is best offered in the context of a multidisciplinary team, which would include experts in genetic counseling, surgery, oncology, social work, nursing oncology, and psychology.
Genetic founder mutations are defined by a genetic mutation occurring in one of the founders of a distinct population that results in a high incidence of that particular genetic mutation in a given ethnic group. The most common founder mutations in hereditary breast cancer are 185delAG, 5382insC in BRCA1, and 6174delT in the BRCA2 gene, found in the Ashkenazi Jewish population.12 These three single mutations account for 10% of breast cancer in the Ashkenazi Jewish population; however, a Jewish woman diagnosed with breast cancer at the age of 40 has a 21% to 30% risk of a BRCA gene mutation.13 It is important to note that 10% of Ashkenazi Jewish woman will have a BRCA mutation that is not one of the founder mutations, so in some circumstances full sequencing of the BRCA genes is recommended.14 Due to these founder genetic mutations, the prevalence of a BRCA mutation in the Ashkenazi Jewish population is 1 in 40.15 This can be compared to the prevalence of BRCA gene mutations in the general population of 1 in 300 to 1 in 800.16 Additional founder mutations have been detected in the Icelandic population and other ethnic groups.17 Due to this phenomenon, the clinician should always assess the patient’s ethnicity on both the paternal and maternal side when obtaining a family history.
Genetic testing should be considered based on the medical history, as well as if the result will affect the clinical management of the patient. It is also important to test a person diagnosed with cancer first. Typically, this result will accurately predict risk for not only your patient but the family as well. If an unaffected family member were to be tested first, then a negative result is uninformative. It would be unknown if there is still a mutation in the family and the patient in question did not inherit it. Since not all the genes have been identified for hereditary breast cancer, a negative result in a high-risk family should be viewed as inconclusive or indeterminate until genetic testing sensitivity increases and can conclusively prove that the patient did not inherit a mutation.
In addition, genetic testing is often complicated by results that reveal a variant of uncertain clinical significance (VUS). This is a common finding occurring in 10% to 13% of Caucasian individuals who have full BRCA gene sequencing, and it occurs at higher rates in minority populations.18,19 These results reveal a subtle DNA sequence change where it is unknown if it is related to a change in protein function. A VUS is commonly a missense mutation or intronic mutation not known to be involved in mRNA processing. For these results, it is important to determine the number of times the particular variant has been detected, if it has been reported with another deleterious BRCA gene mutation, and if it is tracking with the cancer in the family or if additional in vitro studies have been done. Genetic variants that do not track with cancer and are seen with a proven deleterious mutation are less likely to be of clinical significance to the patient.
Hereditary breast and ovarian cancer syndrome (HBOC) is related to mutations in the BRCA1 and BRCA2 genes as well as other undefined genes. Eighty-five percent of HBOC is related to mutations in the BRCA1 and BRCA2 genes.12 These 2 genes have a different clinical presentation, so they will be addressed separately here. However, they are both tumor suppressor genes involved in DNA repair. Over 1000 mutations have been detected to date in both of the BRCA genes. The majority of mutations detected are frameshift, nonsense, or large genomic deletions or duplications that result in premature truncation of the BRCA protein. Since 2006 genetic testing for large genomic rearrangements in the BRCA genes has revealed mutations in an additional 5% of high-risk patients.20
Estimates of cancer risks conferred by mutations in these genes vary according to their ascertainment. The estimated lifetime risk for breast cancer conferred by these genes has been reported to be between 35% and 87%, and the lifetime risk for ovarian cancer ranges from 16% to 60%.14,21-23 The most frequently quoted risk for breast cancer associated with the BRCA genes comes from two studies in 1995 and 1998 by Easton and Ford. These papers give a risk for breast cancer associated with the BRCA1 gene as high as 87% and the risk with the BRCA2 gene as high as 84%.23 It is important to note that these early studies were based on 237 families from the Breast Cancer Linkage Consortium. In order to participate in these initial studies, the research criteria were 4 cases of breast cancer, ovarian cancer at any age, and breast cancer cases at less than 60 years of age—thus these were highly penetrant families. However, a recent meta-analysis of various population-based studies of 18,432 families has estimated the cumulative risk of breast cancer by 70 years to be between 57% and 65% for BRCA1 and 45% and 49% for BRCA2, and the estimated risk for ovarian cancer in these studies was 40% for BRCA1 and 18% for BRCA2.24
The BRCA1 gene is located on chromosome 17q11. It is a large gene with 22 coding exons and 2 noncoding exons. It encodes for 1863 amino acid polypeptide. Most BRCA1 mutation carriers have a “triple negative or basal phenotype” breast tumor profile (estrogen receptor, progesterone receptor, and HER-2/Neu overexpression all negative) with breast cancer that occurs in the third and fourth decade.25 Thirteen percent of breast cancers with a BRCA1 mutation are medullary carcinoma compared to 2% of the general population.26 The breast tumors from BRCA1 mutation carriers tend to be of a higher grade with faster doubling growth rate, especially in younger patients.27 Because of the aggressive nature of these tumors, there is a reduced survival rate for BRCA1 patients. One study revealed that 57% of BRCA1 mutation carriers are diagnosed with breast cancer under the age of 50, compared to 28% of BRCA2 carriers.23 This gene is associated with the highest rate of ovarian cancer (40%).24 Patients with a BRCA1 mutation are reported to develop ovarian cancer at an average age of 38.6. The ovarian cancer that is associated with BRCA1 gene mutations is primarily high grade, advanced stage, and of epithelial origin. Patients with a BRCA1 mutation are reported to be at an increased risk for second breast cancer, ovarian cancer, prostate and pancreatic cancer (Table 8-1).28-31 The risk for a second breast cancer in BRCA1 and BRCA2 mutation carriers is approximately 3% per year or 30% at 10 years.22,28 The risk for prostate cancer in BRCA1 mutation patients is increased only if the proband was less than 65 years of age when determined to have the mutation.29
| Type of Cancer | Reported Risk by Age 70 |
|---|---|
| Breast—initial | 57-65% |
| Breast—second | 3% per year |
| Ovarian | 40% |
| Prostate | None to 2- to 3-fold increase |
| Male breast cancer | 1% |
| Colon | Slight increase |
| Pancreatic cancer | 1-4% |
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


