Genetic Basis of Musculoskeletal Disorders
Sathappan S. Sathappan
Kirill Ilalov
Paul E. Di Cesare
The orthopaedic genome consists of all genes involved in the development and functioning of the musculoskeletal system. The Human Genome Project (1990–2003) has facilitated an in-depth understanding of the orthopaedic genome as well as the molecular biology of musculoskeletal conditions. Current and future molecular interventional modalities are expected to represent a paradigm shift in the management of orthopaedic disorders.
Pathogenesis
Etiology
Human DNA consists of 46 chromosomes (22 pairs of autosomes and 1 pair of sex chromosomes), comprising approximately 30,000 genes.
Each gene represents a chain of nucleotides (Fig. 15-1) characterized by:
Promoter and other regulatory elements that control gene expression
A coding region, consisting of introns and exons, that defines the unique order of amino acids and hence determines protein form and function
Gene expression: the transfer of genetic information (DNA) to messenger RNA by the process of transcription, followed by the translation of mRNA into a specific protein (Table 15-1 gives definitions of terms used in this chapter)
Genetic abnormalities
Can involve individual chromosomal aberrations, such as deletions (as in Turner syndrome), additions (as in Down syndrome), translocations (where portions of two chromosomes are exchanged, as seen in the 11:22 translocation associated with Ewing sarcoma), and nucleotide repeat expansions (as in trinucleotide disorders)
Can affect autosomes and sex chromosomes
Inheritance may be autosomal dominant or recessive.
X-linked mode of inheritance typically is recessive (e.g., Duchenne’s muscular dystrophy, hemophilia) but rarely may be dominant (e.g., X-linked hypophosphatemic rickets).
Many diseases exhibit more than one pattern, depending on the mutation.
Can be inherited or spontaneous (new mutations)
Can follow a simple or complex pattern of inheritance
Single-gene disorders follow Mendelian genetics.
Polygenic disorders result from interaction of multiple genes (e.g., congenital hip dislocation
or neural tube defects, such as myelomeningocele).

Figure 15-1 Outline of the human genome.
Mosaicism refers to an expression of two cell lines in one individual (e.g., X chromosome inactivation in female).
May have variable penetrance (percentage of individuals with the mutation expressing the phenotype) and expressivity (degree to which the individuals with the mutation express the phenotype)
The underlying basis for various musculoskeletal disorders has been attributed to disorders in genotype.
The disease manifestations of abnormalities in the orthopaedic genome (Fig. 15-2) can be broadly classified as:
Skeletal dysplasias
Connective tissue disorders
Neuromuscular disorders
Trinucleotide disorders
Other genetic disorders
Epidemiology
The incidence of many musculoskeletal conditions is increasing due to longer life expectancies. Estimating incidence rates for inheritable musculoskeletal diseases is difficult, since new detection methods, changing classification schemes, and more complete understanding of the disease mechanisms constantly shape our perception of these disorders.
Diagnosis (Algorithm 15-1)
Physical Examination
Age at presentation
Most genetic diseases present early.
Autoimmune disorders with genetic contribution may present in adulthood only.
Presence of characteristic facial features (e.g., Down syndrome and achondroplasia)
Stature: lesser- or greater-than-normal arm spanto—height ratio
Skin lesions (e.g., cafe-au-lait spots)
Neurologic assessment: weakness in myotomes can be either neural or muscle related
Limb configuration and range of motion of joints (e.g., joint contractures)
Laboratory Tests
Hematology (e.g., erythrocyte sedimentation rate elevated in rheumatoid arthritis)
Biochemistry (e.g., creatine phosphokinase [CPK] in Duchenne’s muscular dystrophy)
Molecular diagnostic tests
Gene product analysis using immunoblotting techniques
DNA mutation analysis with polymerase chain reaction (PCR)
Chromosomal studies
Indications include multiple orthopaedic abnormalities, two or more siblings with the same condition, multiple miscarriages in the mother, multiple organ system abnormalities, mental retardation.
Treatment
Varies with specific disease
Supportive
Orthotics to address joint and spinal deformities (e.g., ankle-foot orthosis [AFO] in Charcot-Marie-Tooth disease associated with peroneal muscle weakness)
Physical therapy to condition and maintain tone of functioning musculature (e.g., in spinal muscular atrophy)
Medical treatment
Medications to suppress or curb disease activity (e.g., anti-inflammatory agents and steroids in autoimmune disorders)
Surgical procedures
Fusion procedures
Spinal instability and scoliosis (e.g., for severe atlantoaxial instability in Down syndrome)
Realignment osteotomies
Sofield osteotomies in osteogenesis imperfecta
Lengthening procedures have been considered for short stature in selective cases of skeletal dysplasia.
Table 15-1 Definitions of Terms Associated With Genetic Disorders
Term
Definition
Alleles
Alternate forms of a gene found at the same locus
Anticipation
Phenomenon that refers to a progressive increase in the number of nucleotide triplets down the successive familial generation with worsening in disease presentation
Arthrogryposis
Congenital nonprogressive limitation of joint movement attributable to soft tissue contractures that involve at least two joints
Autoimmune disorders
A group of clinical conditions that have a predilection for accumulation of selective antibodies, the detection of which facilitates clinical diagnosis and categorization
Autosomal dominant
Condition that occurs when a mutation of only one of the alleles is enough to cause the disease, as in achondroplasia, Marfan syndrome, and neurofibromatosis
Autosomal recessive
Condition that requires mutations be present in both alleles of a gene, as in spinal muscular atrophy
Cartilage oligomeric matrix protein
A member of the thrombospondin family of proteins; expressed in high levels in the chondrocyte matrix
Charcot-Marie-Tooth disease
Type 1 or 2 hereditary sensory and motor neuropathy; an autosomal-dominant disease that presents with motor and sensory demyelinating neuropathy
Coding region (of a gene)
Consists of introns and exons; defines the unique order of amino acids and thus determines protein production
Dwarfism
Pathological decrease in stature; can be divided as proportionate (midget) versus disproportionate (short limb or trunk); the short-limb type can be defined by location of maximal shortening as rhizomelic (proximal portion of limb), mesomelic (middle portion of limb), or acromelic (distal portion)
Dysplasia
Intrinsic skeletal developmental abnormality
Ehlers-Danlos syndrome
A group of the most common inheritable connective tissue disorders, consisting of six major subtypes; all have features of skin and joint hypermobility.
Fibroblast growth factors
A group of polypeptide growth factors involved in chondrocyte development and wound healing
Gene
A chain of nucleotides that contain a promoter and a coding region
Hereditary sensory and motor neuropathy
A group of disorders characterized by a slow neural conduction, with muscle biopsy revealing uniformly small-diameter fibers and nerve biopsy revealing demyelination
Mendelian disorders
Disorders involving a single gene
Mosaicism
Expression of two cell lines in the same individual
Muscular dystrophies
A group of inherited, noninflammatory disorders that cause progressive muscular weakness
Neuromuscular disorders
A group of conditions that may have genetic anomalies leading to abnormalities in signal transmission in one of three regions: neural junction, neuromuscular junction, or muscle
Orthopaedic genome
The genes involved in the genesis and functioning of the musculoskeletal system
Osteogenesis imperfecta
A disorder associated with mutation in type I collagen, primarily characterized by bone fragility and long bone deformities
Pleiotropic
A single genotype producing multiple phenotypic effects
Promoter region
The region of the DNA that regulates gene expression
Rheumatoid arthritis
An autoimmune disorder characterized by symmetrical inflammatory arthropathy with varied extra-articular manifestation
Scleroderma
A chronic disease of unknown etiology, characterized by skin fibrosis
Seronegative spondyloarthropathies
A group of conditions that are negative for rheumatoid factor and are associated with inflammatory arthritis and various typical extra-articular manifestations
Skeletal dysplasias
A spectrum of disorders that are caused by abnormalities in bone and cartilage metabolism and phenotypically identified by short stature
Spinal muscular atrophy
An autosomal-recessive disease characterized by loss of anterior horn cells in the spinal cord and presenting with progressive weakness
Transcription
A process of transferring genetic information found in DNA to mRNA
Transduction
The insertion of genetic material into a host cell by use of a viral vector, most commonly an adenovirus
Translation
The process of converting RNA to protein
Trinucleotide disorders
A group of conditions associated with an increased number of nucleotide triplets in DNA
Gene therapy
Gene therapy is a potential therapeutic option when a single functional gene product is absent and treatment is affected by transfer of a wild-type gene; possible orthopaedic examples are Duchenne’s muscular dystrophy, osteogenesis imperfecta, familial osteoarthritis.
Gene transfer into cell lines requires the aid of vectors, which can either be viral (recombinant viruses) or nonviral.
Key issues concerning gene therapy use in orthopaedics
Orthopaedic genetic diseases have an early onset, which necessitates administration of gene therapy at a very early age.
In heterozygous individuals with selective genetic conditions, half of the genes are abnormal and can behave as mutations producing inhibitory protein products that interfere with normal gene products. A successful gene therapy outcome therefore requires eliminating this endogenous mutant allele.
Longevity of gene expression
Economics of gene therapy
Current status of orthopaedic applications for gene therapy
Genetic diseases
Osteogenesis imperfecta
Trials ongoing with implantation of precursors of normal bone (stem cells) into osteogenesis imperfecta mice models
Duchenne muscular dystrophy
In animal models, dystrophin gene delivery success has been hampered by immu-nogenicity and rejection issues.
Nongenetic diseases
Arthritis was the first orthopaedic condition to be targeted by gene therapy.
Involves delivery of antiarthritic genes into synovial linings of diseased joints (rheumatoid arthritis, osteoarthritis)
Phase I trials in human have suggested efficacy in transfer of genes (interleukin-1 receptor antagonists) in patients with rheumatoid arthritis.
Current challenge is to sustain longevity of gene expression.
Osteoporosis
Osteoporotic mice models injected with vector carrying transgenes to block interleukin-1, an osteoclastic cytokine, showed decreased bone loss and in some cases bone mass was restored to normal.
Tissue repair
Gene expression is required for a limited time period until healing is complete.
Bone healing can be enhanced in animal models using an adenovirus that carries the BMP-2 gene.
In experimental models, cartilage defects have been treated using “gene plugs”—bone marrow aspirates combined with a vector carrying a localized “healing” transgene that allows the stem cells to differentiate into chondrocytes and thus repair the cartilage defect.
Skeletal Dysplasias
Definition
Skeletal dysplasias, which are mostly heritable disorders, are caused by abnormalities in bone and cartilage development and phenotypically characterized by a variably short stature.
Epidemiology
The incidence of skeletal dysplasias is 1:3,000 to 5,000 births depending on disease subtype and geographic area.
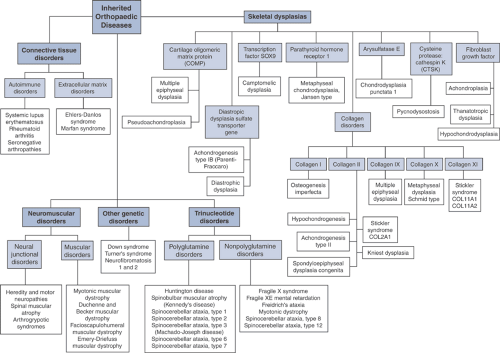 Figure 15-2 Flowchart summary of genetic basis for musculoskeletal disorders. |
 Algorithm 15-1 Diagnostic work-up of musculoskeletal disorders. |
Classification
The clinical classification of dysplasias is based on whether the dwarfism is proportionate or disproportionate, and, if disproportionate, whether it is predominantly short-trunk or short-limb. It is also possible to define these disorders in terms of their genetic basis (Fig. 15-3), as in the section that follows.
Skeletal Dysplasias by Genetic Basis
Skeletal Dysplasias Due to Mutations in Fibroblast Growth Factor Receptor Genes
Fibroblast growth factors (FGF) are a subtype of polypeptides that control cell differentiation and proliferation and are critical in chondrocyte development and wound healing. Mutation in FGF receptor 3 genes leads to:
Abnormal chondrocyte maturation
Impeded endochondral bone growth with rhizomelic shortening (femur and humerus are the most affected as they are the bones with the largest amount of endochondral growth)
Three main disease subtypes
Achondroplasia
Hypochondroplasia
Thanatrophic dysplasia
Achondroplasia
Achondroplasia, an autosomal-dominant disorder, is the most common type of short-limb, disproportionate dwarfism and occurs due to a mutation of guanine to adenine in FGF receptor type 3 (FGFR3) on chromosome 4. Although endochondral ossification is affected, intramembranous ossification is essentially spared. The genetic defect affects the proliferative zone of the physis. The disorder is associated with increased paternal age.
Clinical Features
Frontal bossing, low nasal bridge, midface hypoplasia
Rhizomelic shortening of the extremities (humerus and femur) (Fig. 15-4)
Trident hand: increased space between the middle and ring finger and inability to approximate them
Thoracolumbar or lumbar kyphosis in infancy that usually resolves with the onset of ambulation
Infants are often hypotonic at birth: this can be due to foramen magnum and upper cervical stenosis, which can present with apnea and lead to hydrocephalus. When this is suspected, magnetic resonance imaging (MRI) is indicated.
Radiographs in older children and adults often reveal increased lumbar lordosis with short pedicles and a decreased interpedicular distance, know as “champagne glass” pelvis (width > depth).
The most serious clinical problem is lumbar spinal stenosis, which can be evaluated with computed tomography (CT) or MRI.
Treatment
Fusion for significant cervical instability and decompression for spinal stenosis
Osteotomies for genu varum
Limb-lengthening procedures are a controversial treatment option.
Hypochondroplasia
An autosomal-dominant disorder that results from a cytosine-to-adenine substitution in FGFR3 gene on chromosome 4
The condition, in which mild achondroplasia-type features become apparent at age 2 to 3 years, is characterized by:
Rhizomelic shortening of extremities
Facial features less distinctive than in achondroplasia patients
Lack of trident hand seen in achondroplasia
Neurologic complications such as lumbar stenosis can occur in individuals with hypochondroplasia, but they occur less frequently than in patients with achondroplasia.
 Figure 15-3 Classification of skeletal dysplasias. |
 Figure 15-4 Radiograph of 10-year-old girl with achondroplasia. Note features of rhizomelic shortening of the upper extremity. (From Baitner AC, Maurer SG, Gruen MB, et al. The genetic basis of the osteochondrodysplasias. J Pediatr Orthop 2000;20:594–605.) |
Thanatophoric Dysplasia
Lethal bone dysplasia
New mutations in FGFR3 gene result in new cysteine residues, which trigger the formation of abnormal disulfide bonds between the mutant FGFR receptors.
FGFR receptor complexes then inhibit endochondral ossification.
Clinical Features
Rhizomelic dwarf with large head and normal trunk
Generalized platyspondyly (flattening of the vertebral bodies)
Extreme rib shortening
Skeletal Dysplasias due to Mutations in Collagen Genes
Mutations in Type I Collagen: Osteogenesis Imperfecta
Etiology
Type I collagen (Fig. 15-5) is a major structural protein in bone, skin, and tendons.
A heterotrimer of two identical a-1 chains and one α-2 chain folded into a triple helix configuration
The α-1 and α-2 chains consist of amino acid repeats (Gly-X-Y).
COL1A1 gene, coding for the α-1 chain, is on chromosome 17, whereas the a-2 subunit is coded by the COL1A2 gene on chromosome 7.
COL1A1/A2 gene mutations disrupt the synthesis of type I collagen.
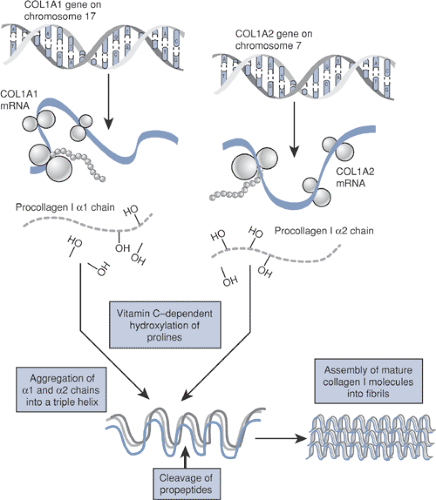
Figure 15-5 Type I collagen synthesis.
Affect all bone and connective tissue containing type I collagen as their major structural protein
Classical findings: bone fragility, long bone deformities (Fig. 15-6)
May also have blue sclerae, dentinogenesis imper-fecta, and scoliosis
Clinical Features and Classification
Most patients are categorized among the four distinct types using the Sillence classification; the rest are classified recently under V, VI, VII types (Table 15-2).
May be autosomal dominant or autosomal recessive. New mutations are rare.
Radiographic Features
Osteoporosis
Multiple fractures
Bowing of long bones (often described as “gracile”)
Deficient ossification of the skull
Diagnosis and Work-up
Prenatal ultrasound and DNA analysis (chorionic villi sampling)
Collagen synthesis analysis of dermal fibroblasts (from skin biopsy)
Osteogenesis imperfecta should be considered in the differential diagnosis of child abuse.
Treatment
Medical treatment options aim to improve bone mass.
Bisphosphonates (pamidronate most common)
Bone marrow transplantation has been used with some success in severe cases but is not standard treatment.
Gene therapy (discussed in detail in section on treatment below)
Bracing may be used for fracture prevention (not effective in management of scoliosis).
Surgical treatment options include Sofield osteotomies
to correct long bone deformities, decompression of basilar impression, and spine fusion for significant scoliosis.
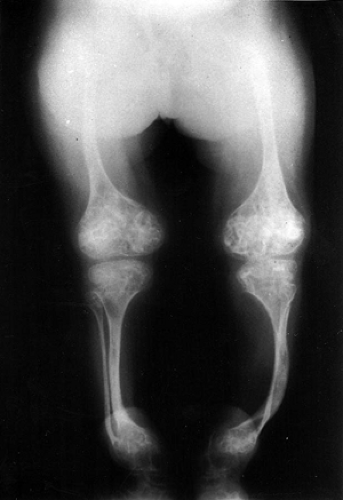
Figure 15-6 Radiograph of a young patient with osteogenesis imperfecta. (From Baitner AC, Maurer SG, Gruen MB, et al. The genetic basis of the osteochondrodysplasias. J Pediatr Orthop 2000; 20:594–605.)Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree

 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

