Introduction
In 1907 Oberndorfer first used the name carcinoid to describe rare ileal tumours with less malignant behaviour than common large bowel carcinomas. Subsequently it became a common name for tumours derived from a widely distributed neuroendocrine cell system, and the carcinoids were classified according to embryological origin into foregut carcinoids (lungs, thymus, stomach, duodenum, pancreas), midgut carcinoids (small bowel to proximal colon) and hindgut carcinoids (distal colon and rectum). The new WHO classification from 2010 introduced the term neuroendocrine tumours (NETs) instead of carcinoid. NETs of the gastrointestinal tract (GEP-NETs) are most common (∼ 60%), followed by NETs of the bronchopulmonary system, whereas other locations (ovaries, testes, hepatobiliary system, among others) have been less frequent ( Table 6.1 ). GEP-NETs are relatively rare, but had increased in incidence to 6.2 per 100 000 in 2005. Since GEP-NETs are overall indolent, their prevalence is high, making them the second most common gastrointestinal (GI) cancer after colon cancer and more prevalent than pancreatic, gastric, oesophageal or hepatic cancer.
| Site | Occurrence (%) |
|---|---|
| Extragastrointestinal (lung, thymic, ovary, uterus) | ∼ 30 |
| Oesophagus | < 1 |
| Stomach | 4–8 |
| Duodenum/pancreas | < 2 |
| Small intestine | 25–30 |
| Appendix | 6 |
| Colon | 10 |
| Rectum | 15 |
Many small-intestinal NETs may be clinically silent and subclinical tumours have been detected at autopsy with an incidence of 8%. Appendiceal NETs have been commonly found in autopsy studies and previous clinical series, although recent reports indicate an increased proportion of small-intestinal, pulmonary, gastric and rectal NETs, mainly due to increased awareness and improved detection.
The recent WHO classification divides NETs in stages according to proliferation rates determined by Ki67/Mib-1 antibody staining (against proliferation antigen) and mitotic index (number of mitoses per 2 mm 2 or 10 high-power fields) ( Box 6.1 ). Grade 1 tumours have a low rate of mitosis and low proliferation rate, Ki67 index ≤ 3%, Grade 2 tumours have Ki67 index 3–20% and Grade 3 tumours Ki67 index > 20%. The most poorly differentiated grade 3 tumours, neuroendocrine carcinoma (NEC), have increased rates of mitoses and higher proliferation index of ≥ 20–40%. The Ki67 proliferation index has become of increased importance in clinical planning. Extensive surgery is more likely to be beneficial for differentiated tumours with low proliferation, in contrast to chemotherapy, which generally has little effect in low proliferating, typicallly small-intestinal NETs.
Chromogranin A and synaptophysin immunostains, which identify proteins of neurosecretory granules, are commonly used to identify NETs. Antibodies against cytosolic markers – neuron-specific enolase (NSE) and PGP9.5 – have also been used for identification of certain tumours, but have generally not been as specific. For poorly differentiated lesions chromogranin staining may be variable and involve only subsets of cells. Synaptophysin reactivity alone, in the absence of chromogranin staining, may indicate an exocrine tumour with endocrine differentiation, or a so-called mixed exocrine–endocrine carcinoma. Dominant secretion (e.g. serotonin, histamine, gastrin, somatostatin) may also be detected in NETs, and occasionally ectopic hormone production is seen, such as adrenocorticotropic hormone (ACTH) and corticotropin-releasing factor (CRF). The latter two sometimes cause an ectopic Cushing’s syndrome in association with NETs ( Table 6.2 ).
| Organ of origin | Hormone production * | Syndrome |
|---|---|---|
| Thymus | ACTH, CRF | Ectopic Cushing’s syndrome, acromegaly, atypical carcinoid syndrome |
| Lung | ACTH, CRF, ADH, GRH, gastrin, PP, hCG-α/β, serotonin | |
| Stomach | Gastrin, histamine (serotonin) | Atypical carcinoid syndrome |
| Duodenum | Gastrin, somatostatin | Zollinger–Ellison syndrome |
| Pancreas | (Serotonin) | (Carcinoid syndrome) |
| Jejunum–ileum | Serotonin, NKA | Classical carcinoid syndrome |
| Prox. colon | Substance P, bradykinin, prostaglandins | |
| Appendix | No hormone production (serotonin) | (Carcinoid syndrome) |
| Colon | PYY | |
| Rectum | CG-α/β |
* ACTH, adrenocorticotropic hormone; ADH, antidiuretic hormone (vasopressin); CRF, corticotropin-releasing factor; GRH, growth hormone; hCG, human choriogonadotropin (α /β subunits); NKA, neurokinin A; PP, pancreatic polypeptide; PYY, peptide YY.
Oesophageal NETs
Oesophageal NETs are exceedingly rare, and occur with male predominance at an age of around 60 years. Most tumours are found in the lower third of the oesophagus or in the gastro-oesophageal junction. Symptoms are non-specific and similar to adenocarcinoma or squamous cell carcinoma, and patients rarely exhibit a carcinoid syndrome. Lymph node metastases have been present at diagnosis in 50% of patients; survival correlates with stage of the disease and overall is poor.
Gastric NETs
Gastric NETs are rare tumours, constituting less than 1% of gastric neoplasms and approximately 8% of all GEP-NETs ( Table 6.1 ). Most of these tumours are derived from enterochromaffin-like (ECL) cells of the gastric fundus and corpus. The tumours are immunoreactive to chromogranin A, synaptophysin and histamine, and can be identified with the specific marker vesicular monoamine transporter isoform 2 (VMAT2). The majority of gastric NETs occur secondary to hypergastrinaemia in patients with chronic atrophic gastritis (CAG) ( type 1 gastric NETs ). These NETs are typically multicentric and develop concomitant with ECL cell hyperplasia in the fundus and corpus, or occasionally in the transitional zone to the antrum. Similar non-antral and multicentric NETs and ECL cell hyperplasia have less frequently been diagnosed in patients with hypergastrinaemia and multiple endocrine neoplasia type 1 (MEN1)-related Zollinger–Ellison syndrome (ZES) ( type 2 gastric NETs ). Both type 1 and 2 gastric NETs develop from ECL cell hyperplasia in a stepwise progression through dysplasia to formation of carcinoid NETs.
More uncommon are gastric NETs that develop as sporadic, generally solitary tumours, without concomitant endocrine cell hyperplasia ( type 3 gastric NETs ). The sporadic NETs are often larger when detected, and more frequently associated with metastases. Like the other gastric NETs, the sporadic tumours most often develop from ECL cells, but may also originate in serotonin-producing enterochromaffin (EC) cells, or contain a mixture with other endocrine cell types. The ECL cells have the ability to secrete histamine, and disseminated lesions of the solitary type may occasionally be associated with an atypical carcinoid syndrome. Exceptionally prepyloric or antral sporadic tumours may produce gastrin and should be named gastrinoma. Another group of gastric NETs consists of poorly differentiated tumours, composed of intermediate-sized or small cells – type 4 gastric NETs ( poorly differentiated NECs ).
Both type 1 and 2 gastric NETs are generally classified as well-differententiated neuroendocrine tumours of grade 1 (benign behaviour) or rarely grade 2 (with uncertain behaviour), whereas type 3 gastric NETs span from well-differentiated tumours of grade 1 or 2 to well-differentiated NEC grade 2. All tumours of type 4 are poorly differentiated NECs of grade 3.
The incidence of gastric NETs has increased, due to more frequent gastroscopic examinations, such as during population screening for gastric cancer, or when screening studies have been performed in patients with atrophic gastritis or pernicious anaemia.
Type 1: gastric NETs associated with chronic atrophic gastritis
These tumours account for 70–80% of gastric NETs ( Table 6.3 ). They occur occasionally in young individuals, but most commonly in older patients, with a mean age around 65 years, and there is a 3:1 female to male predominance.
| Type 1 (70–80%) | Type 2 (6–8%) | Type 3 (15–20%) | |
|---|---|---|---|
| Description | Chronic atrophic gastritis (type A), pernicious anaemia | MEN1-associated Zollinger–Ellison syndrome | Sporadic |
| Tumour site in stomach | Fundus and body | Fundus and body | Fundus, body and antrum |
| Characteristics | Usually multiple polyps (often < 1 cm, occasionally 1–2 cm) | Usually multiple polyps (< 1–2 cm), occasionally larger | Single, solitary (2–5 cm) |
| Histopathology * /gastric fundal biopsy | ECL cell lesion; progression: hyperplasia–dysplasia–neoplasia | ECL cell lesion; progression: hyperplasia–dysplasia–neoplasia | ECL, EC or other cells; normal adjacent mucosa |
| Biological behaviour | Slow growth, rarely metastasise | Usually slow growth, lymph node metastases 30%, liver metastases 10–20% | Aggressive, frequent metastases to regional nodes (71%) and liver (69%) |
| Plasma gastrin | Elevated | Elevated | Normal |
| Acid output | Low or absent | High | Normal or low |
These NETs develop in patients with autoimmune CAG type A, where atrophy of the fundic mucosa is accompanied by pentagastrin-resistant achlorhydria and vitamin B 12 malabsorption. More than half of the patients also have pernicious anaemia ( Table 6.3 ). Reduction of gastric acid and increase in pH stimulates gastrin secretion from gastrin (G) cells in the antrum, and the resulting hypergastrinaemia induces hyperplasia of the fundic ECL cells. Diffuse argyrophilic hyperplasia of the non-antral mucosa occurs in 65% of patients with CAG and micronodular/adenomatoid hyperplasia in 30%, whereas the precarcinoid and dysplastic, enlarged micronodules develop mainly in patients with gross NETs. The tumour development progresses from the specified stages of hyperplasia, through dysplastic stages, to neoplastic intramucosal or invasive NETs.
It is important to emphasise that, although CAG is common in elderly individuals, only few affected patients (1%) ultimately develop gastric NETs. The tumours occur mainly in patients with a long duration of a markedly raised serum gastrin, who have more prevalent hyperplastic changes. The CAG-associated NETs contain predominantly ECL cells, intermingled with other specific or poorly defined endocrine cells.
The type 1 gastric NETs are predominantly located within the body or the fundus of the stomach or in the transitional zone to the antrum. They are frequently multicentric, consisting of multiple, small gastric polyps and invariably associated with ECL cell hyperplasia and microscopic tumours. The number of gross lesions can, however, be limited and some tumours may thus appear as solitary. Individual tumours can present as broad-based, round polypoid lesions, reddish to yellowish, depending on the thickness of the covering mucosa. Some lesions can be flat and broad, or appear as discoloured spots or simply as slight mucosal protrusions. Only a few are ulcerated or bleeding. The number and size vary from innumerable pinpoint-sized tumours to a few or solitary prominent lesions ranging from a few millimetres up to 1–1.5 cm, and only occasionally larger tumours (> 2 cm). The polypoid NETs may be difficult to distinguish from hyperplastic polyps, which are also more frequent in patients with CAG.
Small type 1 gastric NETs are almost always benign with low risk of invasion beyond the submucosa. Larger lesions (> 1 cm) are also predominantly benign but may rarely have invasion of the muscularis propria (< 10%). CAG-associated NETs have a lower incidence of metastases and more favourable outcome than other types of gastric NETs. Metastases to regional lymph nodes occur in 5% and distant metastases in around 2%. However, earlier reports with a greater proportion of large lesions described more frequent metastases. Disease-related deaths are exceptional.
Occasional CAG patients have harboured larger, considerably more invasive tumours, representing poorly differentiated neuroendocrine carcinomas or composite endocrine tumours/adenocarcinoma (see below). All these have an unfavourable prognosis.
Type 2: NETs associated with ZES in MEN1 patients
These gastric NETs constitute 6–8% of all gastric NETs, and are thus much less common than those associated with atrophic gastritis ( Table 6.3 ). They occur in patients with MEN1 and gastrin-producing tumours as a cause of ZES, with equal female:male distribution, at a mean age of 45–50 years.
ECL cell hyperplasia is found in almost 80% of MEN1 patients with ZES, and fundic gastric NETs develop in up to 5–30% of these patients. In addition to the hyperplasia and dysplasia of ECL cells in the fundic mucosa, the oxyntic mucosal thickness is invariably increased, in contrast to the atrophy of type 1 lesions. These NETs also develop in a hyperplasia–dysplasia–neoplasia sequence, but have mainly been associated with diffuse hyperplasia, and less evident micronodular changes. However, gastric NETs in sporadic ZES are rare, and virtually only ZES in association with MEN1 seems to promote growth of gastric NETs. The MEN1 syndrome is caused by an inherited mutation of the MEN1 tumour suppressor gene located on chromosome 11q13. As in other MEN1 lesions, the gastric NETs in MEN1 lose their single remaining functional copy of the MEN1 gene by chromosomal deletions. In MEN1 patients without ZES, gastric NETs are extremely rare. Thus, hypergastrinaemia seems to be required for development of gastric NETs from ECL cell hyperplasia. However, additional factors are obviously needed for tumour formation, since only 1% of patients with hypergastrinaemia due to atrophic gastritis and < 1% of patients with sporadic ZES develop gastric NETs.
The type 2 gastric NETs are located in the gastric body and fundus, and occasionally in the antrum, and are composed mainly of ECL cells with sparse other cell types. They are most often multiple and small (73% are smaller than 1.5 cm), although often larger than type 1 tumours, with a size varying from 0.5 to 2 cm. Occasionally, there are markedly larger tumours. The malignant potential is intermediate between that of CAG-associated gastric NETs and sporadic NETs, and 90% will not have infiltrated beyond the submucosa. However, lymph node metastases are present in up to 30% of the patients, and distant metastases, assumed to originate from gastric NETs, occur in 10–20%. MEN1 patients develop distant metastases from other MEN1-associated tumours as well, and the gastric NETs are not the most likely origin. The overall prognosis will depend more on the other MEN1 lesions, and the prognosis is usually rather favourable for the type 2 NETs. However, rare cases of highly malignant neuroendocrine gastric carcinomas with poor prognosis have also occurred in some MEN1/ZES patients.
Type 3: sporadic gastric NETs
Tumours with no association to hypergastrinaemia account for 15–20% of the gastric NETs and have features that markedly differ from type 1 and type 2 lesions ( Table 6.3 ). They occur sporadically, are usually solitary, and grow much more aggressively. Many are already disseminated at diagnosis. These NETs have a male predominance, with a male:female ratio of 3:1; mean age of presentation is reportedly around 50 years. The sporadic gastric NETs occur in non-atrophic gastric mucosa, without endocrine cell proliferation. Determination of serum calcium and examination of the family history may help exclude the MEN1 syndrome, which should be suspected in all patients with foregut NETs unrelated to atrophic gastritis.
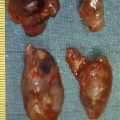
The sporadic tumours are often large; 70% are > 1 cm with a mean size of 3.2 cm ( Fig. 6.1 ). Two-thirds of the lesions will have infiltrated the muscularis propria and 50% invaded all layers of the gastric wall. Some tumours occur in the antral, prepyloric regions, although the majority are located in the body and fundus of the stomach. Most tumours originate in argyrophilic ECL cells, but a mixture of other cell types and EC cells may be present, and are associated with a less favourable prognosis. Regional lymph node metastases have been described in 71% of the patients, and liver metastases have ultimately developed in 69% of the patients. Half of patients are alive after follow-up of 5 years, but patients with distant metastases have a 10% 5-year survival.
The sporadic NETs are generally well differentiated, but often of grade 2 with Ki67 index > 2%. According to previous classification they may have typical or atypical histology, where atypical implies marked nuclear pleomorphism, increased number of mitoses, and areas of necrosis. The atypical tumours are larger, more frequently invasive and commonly associated with metastases at diagnosis. A series of sporadic gastric NETs with atypical morphology reported a mean size of 5 cm and unfavourable survival.
An atypical carcinoid syndrome has developed in 5–10% of the patients with sporadic gastric NETs and is associated with tumour release of histamine. The syndrome is characterised by bright red cutaneous flushing, often with a patchy ‘geographic’ distribution, cutaneous oedema, intense itching, bronchospasm, salivary gland swelling and lacrimation. The atypical carcinoid syndrome is related to histamine secretion, and urinary estimates of the histamine metabolite methylmidazole-acetic acid (MelmAA) may serve as a tumour marker. Most gastric NETs are deficient in the enzyme l -amino acid decarboxylase, and only a few patients have elevated levels of serotonin. Urinary excretion of the serotonin metabolite 5-hydroxyindoleacetic acid (5-HIAA) is therefore less appropriate as a tumour marker. However, the precursor 5-hydroxytryptophan (5-HTP) may be excreted and partly decarboxylated in the kidney, and patients with disseminated sporadic gastric NETs may exhibit some elevated urinary 5-HIAA values.
Gastrinoma
Tumours with sparse staining for gastrin may occur in association with chronic atrophic gastritis. Tumours with intense positive staining for gastrin are rare in the stomach, and are more commonly located in the prepyloric mucosa close to the duodenum. Few gastric NETs cause hypergastrinaemia and peptic ulcer disease, but they still represent an exceptional but possible origin of gastrin excess and ZES. Rare tumours may present with ectopic Cushing’s syndrome due to ACTH secretion.
Poorly differentiated gastric neuroendocrine carcinomas
The poorly differentiated neuroendocrine carcinomas (NECs) are highly malignant neoplasms, with generally extensive local invasion and metastases already at diagnosis. They are not associated with the carcinoid syndrome and occur at a mean age of 60–70 years, with male predominance. Atrophic gastritis has been revealed in half of the patients, but is not believed to cause the tumour, since only a minority of patients have hypergastrinaemia. The majority of tumours are located in the gastric corpus or fundus, but 10–20% may occur in the antrum. The tumours are generally large, with median size of around 4–5 cm, invariably deeply invading the gastric wall and associated with metastases. The majority of lesions appear as ulcerated tumours and a quarter are fungating. They all tend to be of histological grade 3, and often have solid structures with necrosis, a high degree of atypia, frequent mitoses and high proliferation index in Ki67 staining (generally around 20–40%). Almost all tumours show vascular and perineural invasion. In contrast to the well-differentiated gastric NETs, the poorly differentiated NECs may have sparse immunoreactivity to chromogranin A, at least in the majority of tumour cells. Immunoreactivity to synaptophysin (and possibly NSE or PGP9.5) may verify the neuroendocrine differentiation and separate these tumours from exocrine carcinomas. The prognosis is poor with a median survival of 8 months, albeit some individuals are reported to be still alive after follow-up of 10–15 years.
The possibility of progression from well-differentiated gastric NETs, especially type 3 lesions, to NECs has been suggested by a few cases with coexisting well and poorly differentiated gastric NETs.
Aberration of the p53 tumour suppressor gene and chromosomal deletion of the long arm of chromosome 18 are common in poorly differentiated NECs, and occasionally found also in type 3 sporadic gastric NETs. These genetic defects occur in gastrointestinal exocrine carcinomas and may promote aggressive tumour growth. Genetic studies of rare mixed neuroendocrine/exocrine gastric carcinomas indicate that the endocrine tumour component may originate in adenocarcinoma cells.
Clinical evaluation
Symptoms and patient history
The majority of gastric NETs found in elderly patients with atrophic gastritis are detected incidentally by gastroscopy evaluation for anaemia or uncharacteristic abdominal symptoms, or by routine endoscopic screening. Minimal bleeding and anaemia may occur from these NETs, generally the larger, mainly sporadic ones. The largest of these tumours may cause more obvious bleeding, and the poorly differentiated tumours especially may mimic gastric carcinoma, with gastric outlet obstruction. Some become apparent because of metastases, and a few disseminated sporadic NETs present with the atypical carcinoid syndrome.
Patient history should explore the presence of CAG or pernicious anaemia- or MEN1-related endocrinopathies (hyperparathyroidism, endocrine pancreatic or pituitary tumours), both in the patient and family members.
Diagnosis
Diagnosis of CAG type 1 NETs is based on demonstration of high levels of serum gastrin, lack of gastric acid secretion and demonstration of atrophy of the oxyntic mucosa, with concomitant ECL cell hyperplasia in mucosal biopsies from the fundus ( Table 6.3 ). The serum levels of gastrin are also high in patients with MEN1 gastrinomas, although these patients have contrasting high gastric acidity.
Thorough gastroscopic examination should be performed to evaluate multiplicity and size of the gastric NETs. Tumour biopsies should be stained with specific endocrine tumour markers and proliferation markers (chromogranin A, Ki67 staining), and should carefully investigate infiltrative depth and possible vascular invasion. In addition, biopsy samples should be taken from antrum (two biopsies) and fundus (four biopsies) to reveal concomitant ECL cell hyperplasia or dysplasia, the presence of atrophic gastritis, or the contrasting increased oxyntic mucosa thickness of rare MEN1/ZES ( Table 6.3 ). Multiple NET polyps in the gastric fundus in an elderly individual are most likely to represent CAG-associated NETs, since MEN1-associated NETs are rare. A larger, solitary tumour is more likely to be sporadic ( Fig. 6.1 ), and the largest ulcerating and most prominent lesions may be poorly differentiated. Endoscopic ultrasound (EUS) should be added to the gastroscopic examination in type 1 and 2 lesions of size > 1 cm and all type 3 lesions to give information about infiltrative depth. EUS may also reveal associated lesions in the pancreas or duodenum in MEN1 patients, and possibly also show metastases to regional lymph nodes or the liver.
Biochemical screening for other MEN1 endocrinopathies should include analysis of serum calcium and parathyroid hormone, pituitary-related hormones such as growth hormone, prolactin and insulin-like growth factor 1 (IGF-1), and pancreatic hormones ( Box 6.2 ). For cases with the atypical carcinoid syndrome, screening should include urinary analysis of the histamine metabolite MelmAA. Serum values of chromogranin A are generally raised in patients with CAG and ECL cell hyperplasia. These values are also the most important tumour markers, and since they tend to reflect the tumour load, they are often used to monitor patients undergoing treatment for advanced gastric NETs.
- •
Serum calcium
- •
Parathyroid hormone
- •
Chromogranin A
- •
Pancreatic polypeptide (PP)
- •
Gastrin
- •
Insulin, proinsulin/glucose
- •
Glucagon
- •
Prolactin
- •
Somatomedin C (IGF-1)
Computed tomography (CT) with contrast enhancement is routinely performed for tumour delineation and to detect regional lymph node or liver metastases. Scintigraphy using [ 111 In]octreotide scintigraphy (OctreoScan) can often efficiently reveal metastatic spread from differentiated NETs.
Treatment
CAG-associated type 1 gastric NETs
These may disappear spontaneously, and few show more marked progression. Small, multicentric lesions may be followed with annual repeated endoscopy.
Polyps < 1 cm are generally indolent and can be followed by yearly endoscopic surveillance. Tumours > 1 cm without invasion can be treated with endoscopic mucosal resection or multiple band mucosectomy. Few larger invasive tumours require local surgical excision and only rare larger, multifocal lesions need gastric resection. EUS is recommended for evaluation of invasiveness. In cases of malignant development or recurrence despite local surgical resection, partial or total gastrectomy with lymph node dissection is recommended.
Antrectomy has been recommended for treatment of CAG-associated gastric NETs, with the aim of inhibiting antral overproduction of gastrin. The antrectomy is claimed to cause regression of ECL cell dysplasia and small NETs; however, large, invasive or metastatic lesions may remain unaffected. Resection of the antrum may therefore be considered for multicentric or recurrent tumour, and is often combined with surgical excision of larger type 1 NETs, but results and morbidity of this versus repeated endoscopic excision remain unclear.
MEN1-related type 2 gastric NETs
These are more malignant than CAG-associated NETs. Surgical treatment should focus both on removal of the source of hypergastrinaemia and on excision of the gastric NETs. Concomitant exposure of both pancreas and duodenum, via duodenotomy, is done to locate both gastrinomas and possible MEN1 pancreatic lesions, and 80% distal pancreatic resection is most often performed. In type 2 ECLomas > 1 cm local excision is recommended, though endoscopic mucosal resection may be considered if EUS excludes muscularis layer invasion. Gastric resection with regional lymph node clearance is advocated for larger tumours.
Gastrectomy may occasionally be required for very large tumours, although gastrin excess in MEN1 gastrinoma is generally efficiently treated by proton-pump inhibitors.
Sporadic type 3 gastric NETs
These are clearly malignant, with a risk of metastases even for the rare small tumours. Most tumours are large and require operative excision, often performed as gastric resection, combined with regional lymph node clearance. Tumours > 2 cm or those with atypical histology or gastric wall invasion or local metastases are most appropriately dealt with by gastrectomy.
In cases with metastases, tumour debulking of lymph gland and liver metastases may alleviate symptoms of an associated carcinoid syndrome and apparently improve survival. Hepatic metastases may be treated with liver resection, hepatic artery embolisation or chemoembolisation, and radiofrequency (RF) ablation (see below). The somatostatin analogue octreotide may palliate symptoms in patients with the carcinoid syndrome. Chemotherapy is likely to be valuable when the proliferation index exceeds 5% and has response rates of 20–40%. Chemotherapy can be combined with other treatment modalities.
Poorly differentiated NECs
These generally have a dismal prognosis, with a median survival of only 8 months. The tumours are rarely suitable for radical surgery and recurrence should be checked for after gastric resection. However, aggressive surgery together with chemotherapy may be an option to consider, especially in patients with mixtures of well and poorly differentiated tumours.
Duodenal NETs
NETs of the duodenum are rare, and comprise less than 2% of all gastrointestinal neuroendocrine tumours. Duodenal adenomas or adenocarcinomas are much more frequent. However, the endocrine tumours are important to recognise because of a possible association with hormonal or hereditary syndromes and the consequential requirements of treatment. Duodenal NETs are so rare that it is difficult to identify prognostic factors and decide optimal treatment on an evidence base.
Gastrinomas
Gastrin-cell (G-cell) tumours – gastrinomas – are the most prevalent and constitute 60% of the duodenal neuroendocrine tumours; 15–30% of G-cell tumours cause clinical ZES, the remainder are clinically silent. Most gastrinomas are located in the first and second parts of the duodenum. They are frequently small (often around 0.5 cm or smaller), with early metastases to regional lymph nodes reported in 30–70% of patients. The regional lymph node metastases may be considerably larger than the primary tumours, which sometimes may be difficult to detect even at operation. There is generally considerable delay before liver metastases develop, and this is claimed to provide a favourable interval for surgical treatment. It has been recognised that 40–60% of gastrinomas responsible for ZES are located within the duodenal submucosa. MEN1/ZES patients have, in nearly 90% of cases, multifocal duodenal gastrinomas. The duodenal gastrinomas in ZES are slow-growing, indolent malignancies despite their tendency to spread with local lymph node metastases. Duodenal gastrinomas in ZES are rarely identified by endoscopy because of their small size, and they are also often difficult to visualise during surgery. Endoscopic transillumination has been advocated, but it is more efficient to perform a long duodenotomy, allowing discovery of gastrinomas after inversion and palpation of the duodenal mucosa.
Duodenal tumours smaller than 5 mm can be enucleated with the overlying mucosa; larger tumours are excised with full thickness of the duodenal wall. Careful exploration is undertaken for the removal of lymph gland metastases around the pancreatic head. The duodenal gastrinoma is considered as a potentially curable entity of ZES, especially for non-MEN1/ZES. The prognosis is favourable after resection of duodenal gastrinomas in both sporadic and MEN1/ZES patients, with survival reaching 60–85% after 10 years.
Somatostatin-rich NETs
NETs with somatostatin reactivity comprise 15–20% of duodenal neuroendocrine tumours. These tumours are most often clinically hormonally non-functioning. They occur almost exclusively in the ampulla of Vater, causing obstructive jaundice, pancreatitis or bleeding. The tumours appear as 1- to 2-cm homogeneous ampulla nodules, which only occasionally are polypoid, larger or ulcerated. Regional lymph node or liver metastases are present in nearly 50% of patients. Unlike conventional NETs, these tumours have a glandular growth pattern and characteristically contain special laminated psammoma bodies. They can be identified with chromogranin stain. One-third of these lesions are associated with von Recklinghausen’s neurofibromatosis (neurofibromatosis type 1, NF1) and occasionally with phaeochromocytoma. Depending on the size of the tumours and the age of the patient, the somatostatin-rich NETs may be locally excised or removed by pancreatico-duodenectomy.
Gangliocytic paragangliomas
Gangliocytic paragangliomas are rare tumours, occurring almost exclusively in the second portion of the duodenum, and are sometimes associated with neurofibromatosis (NF1). The tumours consist of a mixture of paraganglioma, ganglioneuroma and NET tissue with reactivity for somatostatin and pancreatic polypeptide (PP). The tumours are generally benign, recognised only incidentally or because of bleeding, and have an excellent prognosis following surgical excision.
Other duodenal NETs
More unusual well-differentiated duodenal NETs may display reactivity for other hormones, such as calcitonin, PP and serotonin. Most of these tumours are found in the proximal part of the duodenum as small polyps (< 2 cm). Multiple tumours should raise suspicion of an associated MEN1 syndrome. The majority of these tumours are low-grade malignant and often suitable for local surgical excision. Only rarely do large tumours require pancreatico-duodenectomy.
A distinct group of duodenal NETs without release or staining for hormones is also recognised. These tumours have a somewhat different biology and metastasise less often than the gastrinomas and somatostatinomas. Some of them are asymptomatic and are found incidentally on endoscopic examination. Others present with non-specific abdominal symptoms, gastrointestinal bleeding and sometimes with vomiting or weight loss. Most tumours are located in the first portion of the duodenum, occasionally in the second part and rarely in the third portion (horizontal duodenum). The majority stain for chromogranin A, and some for synaptophysin and/or NSE. Up to one-third of the patients have had other primary malignancies as well, including adenocarcinomas of the gastrointestinal tract, prostate or other organs.
More than half of the tumours are smaller than 2 cm and generally have a good prognosis after resection. Size > 2 cm, invasion beyond the submucosa or presence of mitotic figures are independent risk factors for metastases. Tumours with these risk factors are also likely to recur after apparently curative surgery, even if no lymph node metastases have been detected, whereas lesions smaller than 2 cm rarely metastasise.
Lesions smaller than 1 cm can possibly be endoscopically excised, but re-examination with follow-up endoscopy is required to ensure complete removal. Tumours smaller than 2 cm, without signs of invasion of the muscularis, can be treated by open local excision. The treatment suggested for larger tumours is segmental resection or pancreatico-duodenectomy in order to decrease the risk of recurrence. Periampullary tumours behave in a malignant fashion and need more radical surgery. Patients with metastasising duodenal NETs may survive for decades, substantiating that these NETs are less aggressive than adenocarcinomas.
Duodenal NECs
Poorly differentiated NECs in the duodenum are exceptionally rare. Most occur in the ampulla of Vater, and the patients present with obstructive jaundice and invariably with a rapidly fatal course.
Pancreatic NETs
Pancreatic islet cell tumours are generally classified according to their dominant hormone secretion, or depicted as clinically non-functioning if not associated with any clinical syndrome of hormone excess. Exceptionally, endocrine tumours of the pancreas stain intensely for serotonin (and may also contain other biogenic amines). They appear histologically as classical GEP-NETs, but few have been associated with the carcinoid syndrome. The tumours are managed surgically according to guidelines similar to those for other malignant endocrine pancreatic tumours. Hepatic metastases and a carcinoid syndrome may be treated with somatostatin analogues and interferon, or chemotherapy in the presence of a higher proliferation rate.
Jejuno-ileal (small-intestinal) NETs (midgut carcinoids)
The jejuno-ileal NETs originate from intestinal enterochromaffin (EC) cells in intestinal crypts. They have been named ‘classical’ midgut carcinoids, and typically display serotonin immunoreactivity in the tumour cells. The small-intestinal NETs have increased in frequency, and constitute ∼ 30% of GEP-NETs. Being the most common cause of the carcinoid syndrome, these tumours have often prevailed at referral centres, since this syndrome has required somewhat complicated and often combined medical and surgical treatment. The small-intestinal NETs account for 25% of small-bowel neoplasms and have been diagnosed at an average age of 65 years, with slight male predominance.
Morphological features
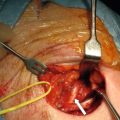
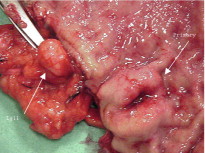
The primary small-intestinal NET is most commonly located in the terminal parts of the ileum, often appearing as a small, flat and fibrotic submucosal tumour, measuring around 1 cm or less ( Fig. 6.2 ), occasionally with some central navelling. Sometimes the tumour is so tiny that it is difficult to detect at surgery, appearing only as a limited area of fibrosis or circumscribed thickening of the intestinal wall. In up to one-third of patients, multiple smaller neuroendocrine polyps occur in the nearby intestine, most likely caused by lymphatic dissemination. In a few cases additional larger neuroendocrine polyps have been found in proximal parts of the intestine, and may appear to represent additional primary tumours. Among patients subjected to surgery, the incidence of mesenteric metastases has been as high as 70–90%, irrespective of tumour size. When growing close to the intestinal wall such metastases have sometimes been mistaken for primary tumours. In contrast to NETs located elsewhere in the gastrointestinal tract, microscopic or gross metastases have also occurred in association with the smallest primary tumours. Unusual large primary tumours sometimes extend directly into a conglomerate of mesenteric lymph gland metastases. The mesenteric metastases typically grow conspicuously larger than the primary tumour, and characteristically evoke a marked desmoplastic reaction with pronounced mesenteric fibrosis ( Fig. 6.3 ). The fibrosis might result from local effects of serotonin, growth factors and other substances secreted from the neuroendocrine metastases.
With more extensive fibrosis the distal ileal mesentery often becomes contracted and tethers the mesenteric root to the retroperitoneum, with fibrous bands attaching to the serosa of the horizontal duodenum. Occasionally, fibrosis and tumour extend over parts of the transverse or the sigmoid colon.
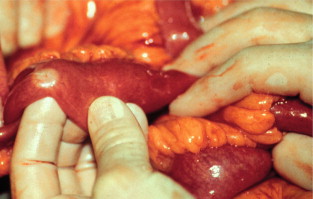
The mesenteric tumour and fibrosis can often cause partial or complete small-intestinal obstruction by kinking and fibrotic entrapment of the intestine, whereas the primary tumour is only occasionally large enough to obstruct the intestinal lumen. Obstruction of the duodenum tends to occur at advanced stages. The mesenteric vessels are often encased or occluded by the growing mesenteric tumour, with resulting local venous stasis and ischaemia in the small intestine, and occasionally frank impairment of the intestinal circulation. Variable segments of the small intestine thus appear dark blue to reddish due to incipient venous gangrene ( Fig. 6.4 ), or occasionally pale and cyanotic due to deficient arterial circulation. A specific angiopathy, called vascular elastosis, occurs with advanced small-intestinal NETs and causes marked thickening of mesenteric vessel walls due to elastic tissue proliferation in the adventitia; this contributes to the intestinal vascular impairment. However, compression by tumour and fibrosis has in our experience been the obvious cause in patients where intestinal ischaemia was revealed at operation.
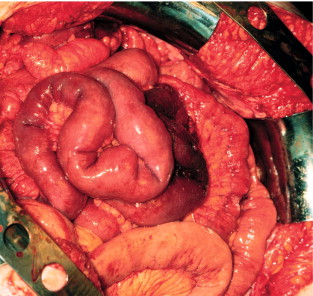
Fibrotic attachments between intestines invariably become more pronounced after surgery and frequently create a conglomerate of distal small-intestinal loops and caecum, which becomes fixed to the posterior and anterior abdominal wall.
Distant metastases from small-intestinal NETs occur most commonly in the liver and the patients then often present with variable features of the carcinoid syndrome. Liver metastases are often bilateral and diffusely spread; approximately 10% of patients have fewer or dominant lesions, sometimes with conspicuous growth of individual lesions. Around 10% of patients present with liver metastases, without mesenteric lesions. Spread to extra-abdominal sites can involve the skeleton (spine and orbital framing are predilection sites), the lungs, CNS, mediastinal and peripheral lymph nodes, ovaries, breast and the skin. A neck lymph gland metastasis can sometimes be the primary clinical sign of a disseminated tumour.
Clinical symptoms
The small-intestinal NETs grow slowly and many patients have experienced long periods of prodromal symptoms before the disease has been clinically recognised. Some patients have had symptoms of borborygmi or episodic abdominal pain, others have had unrecognised features of the carcinoid syndrome, with diarrhoea, discrete flush, palpitations or intolerance for specific food or alcohol. Intestinal bleeding is generally rare with small-intestinal NETs due to the moderate size and submucosal location of the primary tumour. Bleeding has been mainly encountered at later stages, with larger, ulcerating primary tumours, or if mesenteric metastases have grown in the intestinal wall. Such metastases have a special tendency to grow into the horizontal duodenum and sometimes cause bleeding. In other cases bleeding has occurred as a result of intestinal venous stasis.
Intermittent attacks of abdominal pain may initially occur, and increase in frequency until the patient develops obvious subacute or acute intestinal obstruction requiring surgery. In 30–45% of patients the small-intestinal NETs are thus revealed at operation for intestinal obstruction, where the patients often have been submitted to surgery without awareness of the diagnosis. In another 50% of patients the diagnosis becomes evident after detection of liver metastases, and some of the patients initially appear with features of the carcinoid syndrome.
Carcinoid syndrome
The carcinoid syndrome occurs in approximately 20% of patients with jejuno-ileal NETs. Monoamine oxidase activity in the liver can generally detoxify substances released from the intestinal and mesenteric tumours, and symptoms associated with the carcinoid syndrome generally imply that the patient has liver metastases. Occasionally the syndrome may be encountered in patients with large retroperitoneal or ovarian lesions, where secretory products exceed the capacity of detoxification or can bypass the liver and drain directly into the systemic circulation. The syndrome includes flushing, diarrhoea, right-sided valvular heart disease and bronchoconstriction. The aetiology of the carcinoid syndrome is related to release of serotonin, bradykinin, tachykinins (substance P, neuropeptide K), prostaglandins and growth factors such as platelet-derived growth factor (PDGF) and transforming growth factor β (TGF-β), as well as occasionally noradrenaline (norepinephrine).
Secretory diarrhoea is the most common feature of the syndrome, but it may sometimes be mild and non-specific initially. Diarrhoea is often most prevalent in the morning and often meal related. The diarrhoea may, however, have many causes in NET patients, especially when they have been previously operated upon or have reached advanced disease stages. Resection of the distal small intestine may cause moderate diarrhoea due to reduced bile salt absorption; other causes are a short bowel or partial intestinal obstruction. In patients with large mesenteric tumours intestinal venous stasis or ischaemia may often contribute significantly to stool frequency. Severe watery diarrhoea and malnutrition can occasionally occur due to occlusion of main mesenteric veins, which can cause severely oedematous, fluid-leaking intestinal segments of variable length.
Cutaneous flushing generally affects the face, neck and upper chest, and is the most typical feature of the carcinoid syndrome. Flushing may, however, be overlooked, especially in females at menopause. The flush may be provoked by stress, alcohol, certain food, aged cheese or coffee. It is often of short duration, lasting for 1–5 minutes, but may occasionally be prolonged for several hours or even days. Flushing may be severe, and when the flush is long-standing there may be frequent telangiectases and persistent blue-cyanotic discoloration of the skin of the nose and chin.
Heart valve fibrosis , affecting tricuspid and pulmonary valves with plaque-like fibrotic endocardial thickening, is a serious and late consequence of a severe and long-standing carcinoid syndrome. Serotonin and tachykinins may influence the heart and cause fibrosis and valvular thickening with retraction and fixation of the heart valves and subsequently regurgitation and stenosis. As many as 65% of patients with the carcinoid syndrome have tricuspid valve abnormalities, and 19% had pulmonary valve regurgitation in one series. In less than 10%, the pulmonary degradation by monoamine oxidase activity is exceeded and the fibrosis may also involve the left-sided heart valves, and possibly contribute to bronchoconstriction.
The carcinoid heart disease may cause progressive cardiac insufficiency with typically right-sided heart failure and severe lethargy, and used to be an important cause of death in patients with the carcinoid syndrome. However, nowadays, after introduction of somatostatin analogues, patients more often die of progressive tumour disease. Affected patients may require heart surgery and replacement of fibrotic heart valves with prostheses. This operation may lead to substantial improvement, but can be associated with complications, especially in older persons. The heart disease can be diagnosed by echocardiography, which should always be performed prior to major abdominal surgery.
Bronchial constriction as part of the carcinoid syndrome caused by small-intestinal NETs is rare.
Diagnosis
Biochemistry
The biochemical diagnosis of small-intestinal NETs is often based on the demonstration of raised concentrations of the serotonin metabolite 5-HIAA excreted in 24-hour urine samples. The raised 5-HIAA values are specific for small-intestinal NETs, but occur only at advanced disease stages and generally imply the presence of liver metastases.
Determination of plasma chromogranin A is a more sensitive measure, which may be used for the early diagnosis of persistent or recurrent small-intestinal NET disease. Circulating chromogranin A levels reflect the tumour load, and serial measurements have become the most important parameter for monitoring disease spread and to follow results of treatment. The chromogranin values may also predict prognosis. However, it is a non-specific marker for any NET, and false-positive values occur with liver or kidney failure, inflammatory bowel disease, atrophic gastritis or chronic use of proton-pump inhibitors.
Human choriogonadotropin α and β subunits (hCG-α/β) may be predictors of poor prognosis. Exceptionally, NETs secrete enteroglucagon and PP, but PP is a non-specific marker, which may be raised in any patients with diarrhoea.
Pentagastrin provocation test
Pentagastrin injection has occasionally been used to verify the presence of occult small-intestinal NET disease by demonstrating flush and a rise in plasma peptides.
Radiology
The primary small-intestinal NETs are generally too small to be diagnosed with conventional bowel contrast studies . At more advanced disease stages, typical arcading of entrapped intestines with segments of partial obstruction may be visualised, and occasionally signs of chronic obstruction, with thickened bowel wall. In patients with large-bowel symptoms entrapment of the sigmoid or transverse colon may be important to identify prior to surgery. Concomitant colorectal adenocarcinoma has been reported in 10–15%, and it may sometimes be necessary to exclude coexisting rectosigmoidal adenocarcinoma by endoscopy, prior to surgery or during follow-up of a small-intestinal NET.
Computed tomography
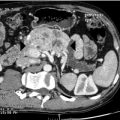
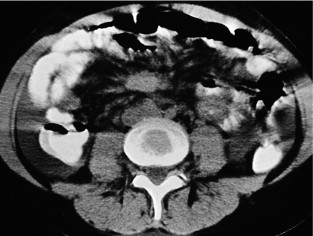
CT can very infrequently visualise a primary small-intestinal NET, but often efficiently demonstrates mesenteric lymph node metastases, retroperitoneal extension of such masses, and liver metastases. The presence of a circumscribed mesenteric mass with radiating densities is very suspicious of a small-intestinal NET with mesenteric metastasis ( Fig. 6.3 ).
Dynamic CT with contrast enhancement is of particular value for planning surgery, when the relation between the mesenteric metastases and the main mesenteric artery and vein is often crucially important to visualise. In cases with advanced intestinal ischaemia CT may reveal a characteristic image of universally dilated peripheral mesenteric vessels and sometimes oedematous loops of intestine. CT with contrast enhancement is often the primary method for visualisation of liver metastases, but may fail to identify the smallest lesions.
Magnetic resonance tomography (MRT) can sometimes be more efficient than CT for demonstration of liver metastases.
Ultrasound
Percutaneous ultrasound is mainly utilised to visualise liver metastases or to guide semi-fine-needle biopsy for histological diagnosis of liver metastases or the deposits in the mesentery. Ultrasound with power Doppler enhancement, and especially use of ultrasound contrast, may increase sensitivity for detection of liver metastases.
OctreoScan®
In nearly 90% of cases, small-intestinal NETs possess somatostatin receptors types 2 and 5, for which the somatostatin analogue octreotide has high affinity. Somatostatin receptor scintigraphy (OctreoScan®) has detected small-intestinal NETs with a sensitivity of 90% and is being increasingly utilised to determine metastatic spread. OctreoScan® is especially efficient for detection of extra-abdominal metastases and can visualise bone metastases better than a routine isotope bone scan, which may miss especially osteolytic metastases.
Positron emission tomography (PET)
PET with the serotonin precursor 5-hydroxytryptophan, labelled with 11 C (5-HTP-PET), or gallium-68 ( 68 Ga) PET can identify the small-intestinal NETs with high sensitivity, and has been used to monitor effects of therapy. PET with [ 18 F]deoxyglucose (FDG) is rarely positive in low proliferating small-intestinal NETs and positivity indicates highly aggressive lesions.
Histology
Needle biopsy specimens from metastases are often used for diagnosis. The NET cells stain immunocytochemically with neuroendocrine tumour markers, mainly chromogranin A and synaptophysin. Reactivity with serotonin-specific antisera implies that a primary tumour should be searched for in the midgut; 85% of jejuno-ileal NETs show reactivity for chromogranin A and serotonin. Proliferation rate is determined with the Ki67 antibody and is most often low in classical small-intestinal NETs (often < 2%).
Most jejuno-ileal NETs show a mixed insular and glandular growth pattern; occasional tumours have a pure insular and trabecular pattern and have been reported as having a slightly less favourable prognosis. Occasional tumours have a higher proliferation rate initially, or during the disease course, and very rarely a lesion may present as an NEC with undifferentiated pattern and very high proliferation rate, with poor prognosis and with sparse effect of surgery.
Surgery
Many patients with small-intestinal NETs will be subjected to acute laparotomy due to intestinal obstruction without suspicion of the correct diagnosis. It is important to appreciate then that the NET is common among small-bowel neoplasms, and findings at laparotomy are often typical with a tiny ileal primary tumour, and conspicuously larger mesenteric metastases with marked mesenteric desmoplastic reaction. The primary tumour and mesenteric metastases should be removed by wedge resection of the mesentery and limited intestinal resection, and lymph node metastases should be cleared as far as possible by dissection around the mesenteric artery and vein and their branches. This procedure is generally indicated even in the presence of liver metastases. If the surgery has been inadequately performed, because the surgeon believed he or she was facing inoperable adenocarcinoma, this will result in the primary tumour and especially the bulk of mesenteric metastases not being removed. Re-operation is then strongly recommended to remove the remaining mesenteric tumour, which may otherwise cause future abdominal complications.
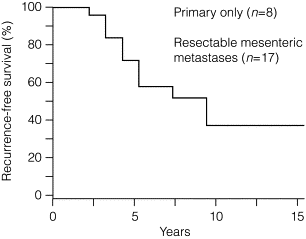
If grossly radical removal of the primary tumour and mesenteric metastases has been accomplished, small-intestinal NET patients may often remain symptom free for long periods. However, small-intestinal NETs are markedly tenacious and recurrence should be looked out for, as the majority of patients (> 80%) will ultimately develop liver metastases if follow-up is long enough ( Fig. 6.5 ). The small-intestinal NETs are unusually slow-growing tumours, and clinically overt recurrence occurs after a median of 10 years and up to 25 years. Earlier diagnosis of such recurrence may be more sensitively based on serum chromogranin A estimates, rather than urinary 5-HIAA measurements.