Abstract
This chapter describes the procedures performed in the in vitro fertilization (IVF) laboratory. Ideal laboratory conditions are discussed to highlight the enormity of the undertaking required to create an environment for optimal clinical outcomes. Procedures performed in the laboratory are introduced in sequence. The conceptual framework underlying each procedure and the details surrounding their execution are delineated. In addition, the current status of preimplantation genetic testing (PGT) is described.
Keywords
IVF laboratory conditions, laboratory procedures, preimplantation genetic testing
Laboratory Environment
- ◆
Conditions in the in vitro fertilization (IVF) laboratory must be tightly regulated to replicate physiologic parameters.
- ◆
The laboratory macroenvironment should be free of contaminants and the air quality rigorously controlled.
- ◆
Establishment of a suitable microenvironment for laboratory procedures involves quality control to maintain a consistent pH and temperature.
In vivo, sperm and egg unite and preimplantation embryos develop within specific, narrowly defined physiologic parameters under sterile conditions. Part of the laboratory’s task is to recapitulate these conditions as closely as possible. Therefore the presence of pollutants can have significant adverse effects on reproductive success. Such contaminants can be microbial, inorganic (i.e., carbon monoxide, nitrous oxide), or volatile organic chemicals (VOCs). They can come from the external environment (e.g., general pollution, construction, industrial hazards) or originate internally (e.g., released from paint, cleaning fluids, flooring, cabinets, and equipment).
Macroenvironment
In order to minimize exposures to contaminants, IVF laboratories must rigorously control air quality. To date, there is no standard practice for the optimization of IVF outcomes. However, IVF laboratories generally have their own central air handling unit (AHU) and/or mobile filter units where the outside (plus recirculating) air is forced through high-efficiency particulate air (HEPA) filters, along with activated charcoal, potassium permanganate, and/or activated carbon filters. The HEPA filters remove particulate matter (i.e., ≥0.3 µm in size) and absorb mold spores and bacteria. The carbon or charcoal filters remove VOCs. In addition, many laboratories have filters, housed within the HVAC ducts, that use chemical/absorbent filtration and/or UV light irradiation to further clean the air and remove microbes and particulates (≥0.01 µm in size), bacteria, and viruses. To maintain the clean air environment, the laboratory should have a high tightness factor (walls and ceilings should have no penetrations), have multiple air exchanges per hour (i.e., 15 to 25 air exchanges/hour), and be under positive pressure to prevent contaminants from adjacent rooms entering the laboratory.
To minimize VOC emissions, all furniture within the laboratory should be stainless steel. The laboratory should have smooth, nonporous walls and lighting should be sealed within special units in order to reduce airborne particles. Inline HEPA and activated carbon filters should be present to remove potential contaminants, such as benzene, isopropanol, and pentane from compressed gas (N 2 and CO 2 ).
A major task for the laboratory is to ensure that a uniform and optimal environment is maintained. It is therefore necessary to use laboratory equipment and media that provide ideal conditions for embryo development: stable temperatures (37°C) and a pH range of 7.2 to 7.4. Fluctuations in temperature have been shown to be detrimental to the oocyte’s microtubular system, causing reductions in spindle size, disorganization of microtubules within the spindle, and, in some cases, complete absence of microtubules. Interference with spindle organization can prevent chromosome disassociation, resulting in aneuploidy. Changes in pH could perturb metabolic homeostasis, stimulate or prevent induction of specific molecular pathways, alter gene expression profiles, and influence embryo development.
IVF laboratories have many pieces of equipment, including incubators, microscopes, lasers, heating stages, water baths, and storage Dewars. There are a variety of options for each piece of equipment; the choice may depend on cost, specific procedures, flow, and size of the laboratory. Incubators may be typical box incubators or bench-top incubators. The incubators may be humidified or dry and/or capable of maintaining low oxygen tension. Stereoscopes may be used for stripping the oocytes, while inverted microscopes with micromanipulators are used to perform intracytoplasmic sperm injection (ICSI), “assisted hatching,” or an embryo biopsy. Regardless of selection, appropriate equipment quality control measures should be performed. An example of a schedule is shown in Table 32.1 .
| Location | Instrument | QC Performed | Frequency |
|---|---|---|---|
| Embryology lab | Incubator |
| Daily *3× weekly Monthly Quarterly Quarterly |
| Isolette |
| Daily Daily Quarterly | |
| Heated stage surface temperatures |
| Daily | |
| Gas room | CO 2 /Nitrogen and mixed gas supply |
| Daily |
| Andrology | Heat blocks |
| Daily |
| Water filter |
| Daily | |
| Computer-assisted sperm analysis |
| Daily | |
| Alarm system |
| Daily | |
| Cryo Lab | Nitrogen Dewars |
| Bi-weekly |
† Viasensor: verification of CO 2 and O 2 partial pressures.
Additionally, quality control testing must be performed on all consumables (e.g., media and plastic ware) that are either newly introduced into the laboratory or are a recurring supply (i.e., a new lot). The purpose of the testing is to make sure that no embryotoxic substances are introduced into the system. This testing is performed using a bioassay (e.g., mouse embryo development, human sperm survival assay) that compares the test results of new material with those of control material that was previously used successfully in the laboratory.
Microenvironment
The media system is a vital component in the process of handling gametes and embryos. The goal is to emulate the in vivo environment as closely as possible. However, the in vivo milieu is dynamic while the in vitro environment is static. Therefore a single ideal culture medium is elusive. The media used in IVF laboratories today consider these and other physiologic issues and provide the most appropriate components at various stages of the IVF process.
Media can be classified based on nutrients (simple or complex) or culture process (one-step, sequential). Numerous studies on embryo metabolism as well as examination of tubal and uterine fluid composition have led to advances in media. There are several complex formulations, each composed of various amounts of substrates including ions, carbohydrates, amino acids, protein, antioxidants, antibiotics, buffers, and chelators. The media system used by the laboratory is dependent on the overall culture system and clinical outcomes.
Media can also be classified based on the buffering system in use. As mentioned previously, the pH of the media is critical to embryo development. The pH of the surrounding environment influences sperm function, embryo metabolism, and organelle localization. Safety concerns and outcomes with respect to human embryo development have narrowed the buffering systems in use within the IVF laboratory. The buffering systems most utilized depend on the particular purpose and the equipment available ( Table 32.2 ). Typically, a sodium bicarbonate buffered based medium is used for culture purposes because detailed studies have shown superior embryo development in the presence of carbon dioxide (CO 2 ) versus room atmosphere. The pH is maintained by way of gas equilibrium; therefore, the equipment must have CO 2 sensors and adjust the amount of CO 2 pressure to maintain the desired pH unique to the medium. A medium that is sodium bicarbonate–controlled and remains in room atmosphere (outside of a CO 2 -controlled environment) even for a short time is susceptible to significant pH changes. Therefore, for procedures performed in room atmosphere, the inclusion of a zwitterionic buffer such as MOPS (pH = 6.5 to 7.9; pK a 7.0 to 7.3) or HEPES (pH = 6.8 to 8.2; pK a 7.2 to 7.5) is required. Both are effective and have been shown to work well clinically. This nonbicarbonate medium is used mainly for procedures such as oocyte collection, oocyte stripping, ICSI, and biopsy. Alternatively, laboratories may have isolettes where CO 2 can be supplied and controlled and a bicarbonate buffer system can be used.
| Media | Type | Sperm Washing | OR | ICSI | Embryo Transfer | Cryopreservation | Embryo Culture | D3 Biopsy | D5 Biopsy |
|---|---|---|---|---|---|---|---|---|---|
| HEPES | Zwitterion based buffer | Yes | Yes | Yes | Yes | Yes | No | Yes * | Yes |
| MOPS | Zwitterion based buffer | Yes | Yes | Yes | Yes | Yes | No | Yes * | Yes |
| HEPES/MOPS mixture | Zwitterion based buffer | Yes | Unknown | Yes | Unknown | Unknown | No | Yes * | Yes |
| Bicarbonate/CO 2 | Bicarbonate based buffer | Unknown | Yes † | Yes * † | Yes † | Unknown | Yes | No | No |
* HEPES or MOPS without Ca 2+ /Mg 2+ is used for D3 embryo biopsy, which allows for easier blastomere removal.
In addition to the environment itself, laboratories must be staffed adequately with properly trained personnel. The work in the laboratory is extremely labor-intensive. Laboratory personnel must have discipline, be detail-oriented, and function as a team. The number of staff depends on the number and types of procedures performed. An IVF lab that is performing all procedures requires a minimum of two embryologists, with an additional embryologist for every 100 to 150 cycles. Each laboratory procedure is standardized. Embryologists follow the standard operating procedures (SOPs) created for every procedure performed in the laboratory. Adherence to the SOPs ensures consistency and reduces systematic errors. As part of the quality assurance/control system, analytics are periodically performed and each embryologist participates in internal and/or external quality assurance programs for each procedure.
Procedures
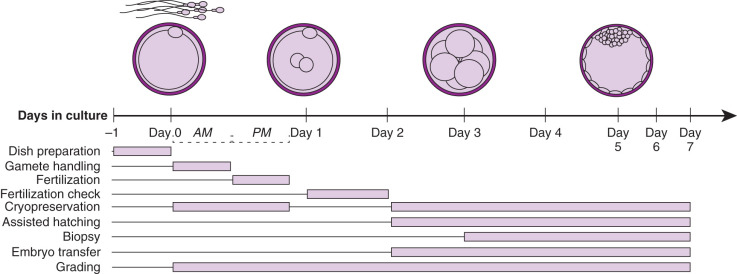
The procedures performed in the laboratory are specific to the developmental stage of gametes and embryos ( Fig. 32.1 ). Additionally, embryologists must coordinate efforts so the timing of events is performed precisely. The following sections cover the concepts underlying and a detailed description of common procedures.
Gamete Handling
- ◆
Oocytes are isolated from follicular fluid (FF) at retrieval and assessed for morphologic appearance. An expanded cumulus-oocyte complex (COC) is suggestive of oocyte maturity.
- ◆
Cumulus cells (CCs) surrounding the oocyte are denuded for ICSI, allowing for assessment of the zona pellucida (ZP), perivitelline space, and oocyte cytoplasm in addition to evaluation of nuclear maturation by detection of the presence (or absence) of a germinal vesicle (GV) and polar body (MII).
- ◆
Ejaculated sperm are processed by a simple wash, migration (swim-up), or density-gradient technique.
- ◆
Surgically retrieved sperm are processed according to the method whereby the sample was obtained: epididymal samples by density gradient and testicular samples by mechanical separation and dissection.
Oocyte Collection
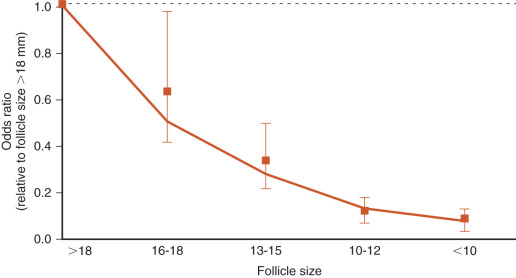
The egg harvest entails aspirating follicular contents from antral follicles. The FF is transferred to the laboratory for evaluation. Approximately 75% to 80% of the follicular aspirates will contain an oocyte. The size of the follicle dictates the likelihood of oocyte recovery and the likelihood that it is mature ( Fig. 32.2 ). Nuclear and cytoplasmic maturation of the oocyte is critical for normal fertilization and embryo development.
In order to prepare for an oocyte collection, each individual case must be prepared for in advance. The day before retrieval, culture dishes are made and equilibrated under controlled conditions with a constant pH and stable temperature at 37°C (see previous text).
Once the FF has been collected, it is passed to the IVF lab where the oocytes are identified using a stereomicroscope and heated stage, usually at 8 to 60× magnification. The embryologist pours the contents of each tube into a petri dish, forming a thin layer of fluid, which can quickly be scanned for the presence of an oocyte. The oocytes are then washed in bicarbonate-based culture medium, transferred into preequilibrated medium, and stored in a CO 2 environment (i.e., a CO 2 incubator) for incubation until further processing.
Assessment of the Oocyte
Techniques that assess the nuclear and cytoplasmic maturation of the oocyte fall into two major categories: invasive and noninvasive. The common practice in embryology laboratories includes a noninvasive morphologic assessment using light microscopy. Each oocyte is assessed for nuclear maturation and morphologic characteristics; this includes evaluating each of the anatomic components of the oocyte (cumulus, ZP, perivitelline space, polar body, and cytoplasm) ( Fig. 32.3 ). The morphologic features of the oocyte have been studied as factors that may be associated with pregnancy outcomes. However, a consensus on whether each morphologic characteristic is associated with outcome has been difficult because many investigators have used different terminology to define similar observations. Although no consensus exists, it is common practice to record these observations. An oocyte scoring system has been developed that takes into account all morphologic characteristics.
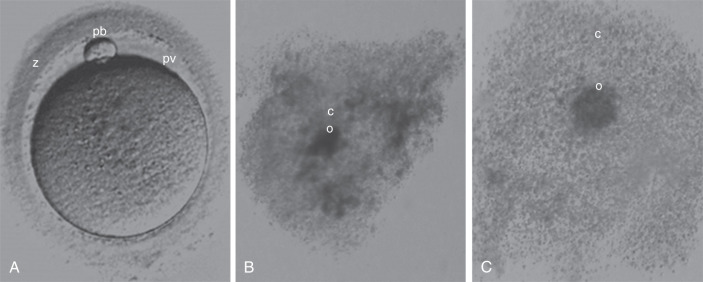
Oocytes collected at retrieval are difficult to visualize because they are surrounded by a cumulus mass composed of follicular cells in a polymerized matrix of hyaluronic acid ( Fig. 32.3B and C ). Therefore, in the case of conventional insemination (CI; see further on), the morphologic assessment is limited to the cumulus-oocyte complex (COC). The volume, density, and expansion of the CCs can suggest the stage of oocyte nuclear maturity. The grading of cumulus expansion is a visual inspection of whether the cells are compact or expanded. Compact CCs take on a darker appearance due to the dense association of these cells around the oocyte (see Fig. 32.3B ); they are usually associated with immature oocytes, while expanding or fully expanded CCs are usually associated with mature oocytes (see Fig. 32.3C ).
Unlike CI, in which intact mature COC are inseminated, fertilization with micromanipulation (ICSI) requires denudation of oocytes (i.e., removal of the surrounding cumulus and corona cells). The denudation allows for an additional detailed evaluation of each component of the oocyte ( Fig. 32.4 ). Starting from the outermost layer, the oocyte is encased in a thick glycoprotein shell called the zona pellucida (ZP). The ZP normally averages 15 to 20 µm in width and is intact and translucent in a mature oocyte (see Fig. 32.3A ). Studies have evaluated whether the darkness and/or thickness of the ZP is associated with clinical outcome. Although darkness has shown little correlation with clinical outcome, the thinner ZP has been associated with higher fertilization rates. Other reported alterations of ZP morphology, including fractured or broken ZP, have not been demonstrated to be associated with clinical outcomes. The etiology of these findings may result from postmaturity of the oocyte or excess pressure during oocyte aspiration.
The space between the ZP and oolemma is called the perivitelline space. Although several studies have suggested that an enlarged perivitelline space ( Fig. 32.5F ) is associated with poorer fertilization and embryo development, there is no clear consensus. An enlarged perivitelline space has been hypothesized to relate to oocyte postmaturity. Sometimes the perivitelline space may contain granules, which do not appear to influence oocyte or embryo development. The first polar body appears in the perivitelline space. Unusually large polar bodies may indicate disturbances in the position of the meiotic spindle, which may occur during oocyte aging. Accordingly, a large polar body has been associated with poor embryo quality and low pregnancy rates. Fragmentation of the first polar body has not been consistently identified as an abnormality.
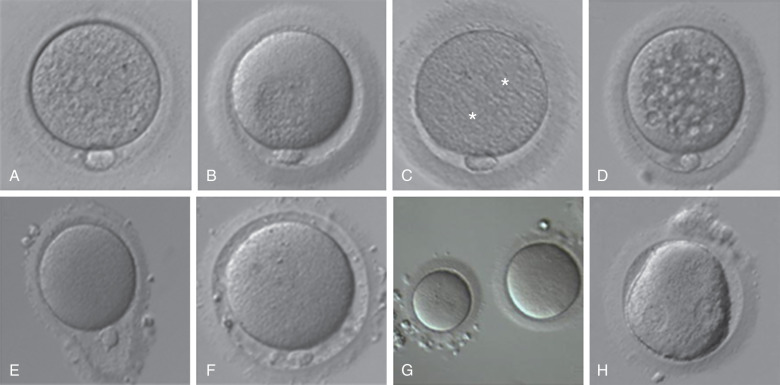
The cytoplasm of a normal egg is typically uniform, translucent, and free of inclusions. A relatively common abnormality that can be visualized by light microscopy is central granularity (see Fig. 32.5B ). This granular area likely represents a polarized distribution of mitochondria and may have implications for cytoplasmic maturity. Studies evaluating granularity, either generalized or central, have failed to demonstrate a consistent association with clinical outcome. Although some studies have shown that cytoplasmic darkness is associated with a poorer fertilization rate or embryo development, others have not. It has also been hypothesized that localized dark and granular cytoplasm may be related to atresia.
The presence of vacuoles, an aggregation of smooth endoplasmic reticulum (SER) within the cytoplasm, has been associated with decreased fertilization, embryo quality, and ongoing pregnancy rates (see Fig. 32.5C ). Vacuoles may be associated with oocyte aging and degenerative processes in oocytes. Case reports have suggested that the presence of SER is associated with an increased risk of Beckwith–Wiedemann syndrome, diaphragmatic hernia, multiple malformations, and ventricular septal defect. A recent study reported healthy live births from embryos derived from such oocytes and recommended a careful follow-up of the children born. As a result, it is recommended that this cytoplasmic abnormality be noted. Inclusions, or refractile bodies, are inconsistently associated with a compromise in embryo quality and pregnancy outcomes. Although their cellular basis is unknown, the presence of inclusions within an oocyte may be associated with atresia.
Various shapes and sizes of the oocyte have been observed. A normal human oocyte is roughly 120 µm in diameter. Oocytes with a diameter of 200 µm or more are commonly called giant oocytes (see Fig. 32.5G ). These oocytes are thought to be giant due to errors in mitosis during proliferation of the oogonia. As a result, these oocytes are likely diploid and the resultant embryos are at increased risk for dygynic triploidy, mosaicism, or polyploidy. The presence of a giant egg in a cohort of oocytes is not associated with overall decreased pregnancy outcomes, but some data have shown that embryos derived from the cohort may show an increased incidence of abnormality in the cleavage rate.
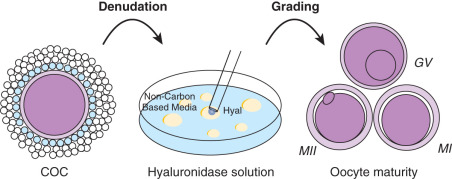
Microscopic observation of denuded oocytes allows for assessment of the presence or absence of a GV. Fig. 32.6 shows three stages of oocyte maturation. On average, 3.9% of the intact oocytes are at the metaphase I (MI) stage, having undergone breakdown of the GV but not extrusion of the first polar body. Approximately 10.3% of the intact oocytes are at the GV stage and approximately 85.8% are in the MII stage, defined by the presence of the first polar body. ICSI is carried out only on MII oocytes, because only such oocytes have reached the haploid state and thus can be fertilized normally. It has been reported that 74% of the MI oocytes complete meiosis within 20 hours after retrieval. Another study reported that 27% of MI oocytes extruded their polar bodies within 4 hours of egg retrieval. Oocytes from this study were injected on the day of the egg retrieval in parallel with the oocytes retrieved at MII. This study demonstrated that MI oocytes that completed their maturation in vitro had lower fertilization rates compared with those aspirated at MII. However, no differences were observed in embryo quality between the two groups of oocytes. Additional studies support these observations, showing that oocytes matured in vitro can be normally fertilized; however, embryos derived from these oocytes rarely result in pregnancies. These results suggest that rescue of MI oocytes of patients with few MII oocytes may increase the number of embryos for transfer; but the chance of improving pregnancy rates by this procedure is minimal. GV-stage oocytes require overnight (24 to 48 hours) incubation in culture media supplemented with gonadotropins in order to reach the MII stage. Few pregnancies have been reported from oocytes that were retrieved at the GV stage, although standard IVF treatment with controlled ovarian hyperstimulation was performed. Because of the poor results, GV oocytes are usually discarded. Denuded and rinsed oocytes are incubated until the time of microinjection.
Polscope imaging provides valuable information on the structure and architecture of the meiotic spindle. The integrity of the meiotic spindle in MII oocytes is crucial for normal fertilization and subsequent development. Overall, meiotic spindles can be detected in up to 91% of human MII oocytes at the time of ICSI. Studies have shown the presence of a birefringent spindle in a human oocyte can predict not only a higher fertilization rate but also greater embryo developmental competence. High degrees of misalignment of the meiotic spindle and the first polar body have also been documented and are thought to occur as a result of polar body displacement during cumulus corona removal. The relative position of the spindle within the oocyte, however, has little influence on the developmental potential of the resulting embryos.
Because most oocytes possess spindles, the mere presence of a spindle is likely of limited value. Whether quantitative spindle analysis (estimating the density of tubules within the spindle) offers added value remains to be seen. So far, visualization of the spindle and the appearance of the first polar body are accurate indicators of oocyte maturity and may help to determine the timing of ICSI (see ICSI ).
The developmental competence of the oocyte has also been assessed through a biomarker approach. Several strategies have been applied, including a comparison of transcriptomes, proteins (proteomics), or metabolites (metabolomics) of granulosa cells (GC) and/or CCs or FF. Evidence suggests that CCs from follicles of embryos that result in a pregnancy may have differential expression. Others have shown that gene-expression profiling in either GC or CC has no association with fertilization outcome but may be associated with advanced maternal age. Estes et al. found that the FF proteome in women 32 years of age or younger predicted ovarian response and live birth. Another study reported that oocytes that resulted in pregnancy presented high amounts of proteins, and those oocytes that resulted in no pregnancy presented high amounts of ubiquitinated peptides. Several investigators have demonstrated associations between FF metabolome and the developmental competence of the oocytes. Although intriguing, the available testing is not ready for routine clinical use. However, given that they represent cellular regulatory processes, biomarkers may be more informative in the future than morphology.
Sperm Collection
Sperm are obtained either through ejaculation (i.e., masturbation, electroejaculation) or by surgical retrieval. The evaluation and assessment of semen is very important for the diagnosis of infertility and for treatment decisions. The collection method for IVF is dependent on the clinical scenario (see Chapter 31 ). The collection processing procedures and assessment of the sperm depend on the method of collection and are discussed in the following paragraphs. The choice of sperm preparation technique depends on the nature of the semen sample, particularly with respect to motile count, ratio between motile and immotile count, volume and presence of antibodies, agglutination, and pus cells or debris.
The majority of sperm collections are obtained through masturbation into a collection cup. The container to collect the sample has to be wide necked and plastic or sterile glass, in addition to being cytotoxically tested. It must be labeled with the patient’s full name, identification number, date of birth, date and time of collection, and number of days of abstinence. Semen samples are collected after 2 to 10 days of abstinence. A shorter period of time may be associated with lower volume but higher motility, while longer abstinence can be lead to reduced sperm motility. In cases of male infertility, shorter durations of abstinence (1 day) may be associated with higher motility and better morphology. The use of lubricant (other than mineral oil or selected lubricants) is discouraged because they may be spermicidal. Other collection methods include the use of Silastic condoms during intercourse.
Most of the samples should be collected on site in a private room set aside for this purpose. The room can be adjacent to the lab and have a pass-through for dropoff; alternatively, the sample can be given to a staff member with proper chain of custody (for specimen verification) protocol. If for personal reasons the patient cannot produce on site, he can produce at home or at a place nearby that will allow sample delivery to the laboratory within an hour. Samples should be kept at room temperature during transport (20 to 37°C) and can stay at room temperature in the laboratory for up to 1 hour in order for liquefaction of the semen to occur.
Assessment of Ejaculated Sperm
Prior to processing the sperm for IVF, a semen assessment is performed, which entails a sperm count and motility evaluation ( Table 32.3 ). The assessment dictates how the sperm is processed and the procedure type for insemination. Sperm counting and motility assessment are obtained through standard procedures. The goal of preparation of sperm is to concentrate the motile spermatozoa in a fraction that is free of seminal plasma and debris. The sperm preparation methods attempt to mimic the natural process whereby viable sperm are separated from the seminal plasma and other constituents as they migrate through the cervix. The seminal plasma contains substances that inhibit capacitation and prevent fertilization. Sperm processing should be performed within 30 minutes if liquefaction is completed or no later than 1 hour after production. If delayed, the uncontrolled production of reactive oxygen species (ROS) that exceeds the antioxidant activity of seminal plasma will lead to oxidative stress and result in decreased sperm capacity. The ROS are produced from leukocytes and other debris present in the seminal plasma. After sperm processing, the insemination process ideally should be performed within 4 to 6 hours. After 4 to 6 hours, sperm DNA fragmentation may be increased. Several sperm preparation techniques are described in the following paragraphs.
| Semen prameters | WHO, 2010 ‡ | 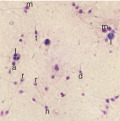 Strict morphology analysis: Observed morphologies in sample include (a) large acrosome, (d) tail defect, (l) leukocyte, (m) macrocephalic head, (n) normal morphology, (r) round head, (t) tapered head. |
| Volume | 1.5 mL | |
| Concentration | 15 × 10 6 /mL | |
| Total sperm concentration | 39 × 10 6 | |
| % Motile | 40% | |
| Progressive motility * | 32% (a+b) | |
| Vitality † | 58% | |
| Morphology (Kruger strict) normal forms, % | 4% | |
| Leukocyte count | <1 × 10 6 /mL |
* a, rapid progression (>25 µm/s); b, sluggish (5–25 µm/s).
‡ World Health Organization, Department of Reproductive Health and Research. Who laboratory manual for the examination and processing of human semen, 5th ed. 2010. Available at: http://www.who.int.easyaccess1.lib.cuhk.edu.hk/reproductivehealth/publications/infertility/9789241547789/en/ .
Processing Techniques
Simple Wash
A simple washing procedure provides the highest yield of sperm and is adequate if semen samples are of good quality. The procedure entails adding culture medium that is a balanced salt solution supplemented with protein and an appropriate buffer. The semen sample is mixed with the buffered medium and centrifuged. The pellet is then resuspended in the medium and centrifuged again. The centrifugation speed (i.e., 300 g ; 1000 rpm) and number of washes should be minimized to avoid the production of ROS within the pellet. The generation of ROS may reduce sperm function and motility, cause peroxidation of sperm plasma membranes, decrease oolemma binding and fusion, and result in failures in fertilization. The number of washes to remove seminal plasma can be reduced by increasing the volume of medium in each tube. Although quite successful, a simple wash alone for IVF is rare due to persistent debris (i.e., leukocytes, epithelial cells), and nonviable sperm that are present in the pellet, which may inhibit capacitation and the ability of the viable sperm to fertilize the oocyte.
Migration: Swim-up
Motile sperm can be further selected by their ability to swim out of seminal plasma and into culture medium. The swim-up is a very common sperm processing technique for IVF. There are multiple ways to perform a swim-up procedure. It involves first washing the semen sample, followed by placing semen underneath a small volume of culture medium (between 0.5 and 1 mL). After 1 hour of incubation (swim-up time) the supernatant is removed. It will contain highly motile sperm (more than 90% motile spermatozoa) and is resuspended in a small volume of culture medium PRIOR to being used directly. This technique is simple and cheap and does not require sophisticated equipment or highly specialized skills. It is most often used for “normal” samples (average or good sperm concentration and motility) given that it has a low yield.
Density Gradient
A density gradient separates sperm based on their density. Although the initial preparation was removed from the market because of possible contamination of endotoxins, there are currently several types of density media based on silane-coated silica particles that have been proven to have very low toxicity. Density gradients can either be continuous or discontinuous. The type of gradient is based on whether there is a continuous gradient from the top to the bottom of the tube or if there are a number of layers of decreasing densities placed on top of one another. Numerous commercial products are available. The most common gradient used for sperm processing in IVF is discontinuous, where a volume of a low (40% to 45%) suspension is layered over a high (80% to 90%) suspension of silane-coated silica particles. The semen sample is placed on top of the suspension and centrifuged. This procedure is based on the principle that morphologically normal sperm, abnormal sperm, and debris have different densities. As a result of the differences in density, normal motile sperm cells penetrate the higher densities in direction of the centrifugation force, whereas immotile or abnormal morphologic sperm are retained at the boundaries of the interphases. This technique isolates the subpopulation of sperm with the best motility, morphology, and nuclear and mitochondrial DNA quality. Density gradient processing of sperm has been associated with the selection of high-quality sperm and higher assisted conception rates than swim-up. The density gradient is the most widely used method for sperm processing after masturbation or electroejaculation in IVF laboratories.
Glass Wool Filtration
Glass wool filtration separates motile sperm from immotile sperm, debris, and leukocytes prior to centrifugation. The process involves removing the debris from the ejaculate through mechanical retention and adhesion to glass fibers. Since the whole ejaculate is filtered, ejaculates from patients with oligozoospermia can be processed. This technique eliminates up to 90% of leukocytes contaminating the semen and therefore reduces ROS significantly. After the filtration process, the semen is centrifuged to remove the seminal plasma. Glass wool filtration is an easy technique to perform and results in the recovery of sperm with good motility.
Magnetic Activated Cell Sorting
Magnetic activated cell sorting (MACS) separates apoptotic from nonapoptotic sperm on a molecular level. Apoptotic sperm externalize phosphatidyl serine residues, which bind to annexin V. The process entails mixing a semen sample after double-density gradient centrifugation with superparamagnetic beads that are conjugated with specific antibodies to annexin V for 15 minutes. The mixture is loaded on a separation column, which is placed in a magnetic field. The nonapoptotic sperm (annexin V–negative) do not bind to the beads and pass through the column. The fraction of the sample that does not bind has better morphology and higher fertilization potential than sperm separated by density gradient alone. Although some clinical studies have shown improvement in cryosurvival and pregnancy rates with the addition of MACS for sperm selection, others have not. More studies are needed before MACS is routinely used for sperm selection in clinical laboratories.
Sperm Stimulation—Pentoxifylline
The goal in any ICSI procedure is to use spermatozoa that are viable. Sperm motility is the best indicator that both the functional (protective) plasma membrane and the metabolic processes are in place. Spermatozoa retrieved from the testis are in a different physiologic state than sperm that have been transported through the epididymis. Many times these sperm (fresh or frozen-thawed) have extremely low motility or are immotile, making it difficult to identify viable sperm for injection. Sperm motility can be stimulated with a variety of chemicals. One of the most commonly used chemicals for stimulating sperm motility is pentoxifylline. It can be administered orally for 3 to 6 months or directly applied in solution to processed sperm and incubated for 1 hour prior to use. Pentoxifylline is a nonspecific inhibitor of phosphodiesterase; it has stimulatory effects on sperm motility. This effect is attributed to elevated intracellular levels of cyclic adenosine monophosphate (cAMP) via inhibition of its breakdown by cAMP phosphodiesterase. Pentoxifylline is also reported to enhance the acrosome reaction due to the increased levels of cAMP. Overstimulation of sperm with pentoxifylline can induce a premature acrosome reaction. Therefore this chemical should be used only on a limited basis where no motile sperm can be readily identified.
Novel Techniques
Some novel techniques are now being assessed to aid in sperm preparation by reducing the presence of ROS. The introduction of antioxidants in the sperm preparation media or removal of the centrifugation step may limit ROS levels. Electrophoretic sperm isolation and microfluidic sperm processing isolate sperm from the seminal plasma without centrifugation and are being investigated as methods of preparation that minimize ROS. It is unknown whether these techniques will improve clinical outcomes.
Surgical Sperm Retrieval
Several sperm retrieval methods have been developed to retrieve sperm from the epididymis and/or testes in men with azoospermia. The technique employed depends on the clinical scenario (discussed in Chapter 31 ). The evaluation and processing of the sample is dependent on the surgical technique and site of collection. Sperm retrieval should be carried out either on the day of oocyte retrieval or the day before, depending on the laboratory workload. Additionally, the surgically retrieved specimen may be incubated for up to several days if the sample is poor and requires further maturation. All surgical collections require ICSI for optimal fertilization (see further on).
Collection, Processing, and Assessment of Surgical Sperm
Epididymal Sample
The procedure to collect sperm from the epididymis is either a microsurgical epididymal sperm aspiration (MESA) or percutaneous epididymal sperm aspiration (PESA). PESA is a blind procedure. The surgical approach is dependent on physician preference and the clinical scenario. A fine needle is used to puncture the epididymis, and the epididymal fluid is collected by fine-gauge needle aspiration. MESA is an open surgical sperm retrieval procedure that uses an operating microscope to locate the tubules of the epididymis. Once they are located, the tubules are opened and the spermatic fluid that flows out is aspirated. Regardless of the approach, the epididymal fluid is placed onto a petri dish and examined under the inverted microscope (400× magnification) to confirm the presence of motile sperm. In most cases, the amount of debris is less than the ejaculated sample and contains a high concentration of motile sperm (1× 10 6 sperm/µL). The preparation technique to isolate the motile sperm is based on the sperm density and motility and the amount of cellular debris. A density gradient is usually used unless the sperm density is low and a simple wash is employed.
Testicular Sample
The procedure to collect sperm from the testes may either be testicular sperm aspiration (TESA) or testicular sperm extraction (TESE). TESA samples are obtained with a wide-bore needle pushed percutaneously into the testis. TESE is an open technique that removes several pieces of testicular tissue. TESA samples are evaluated for motile or immotile spermatozoa with a stereomicroscope. Using fine-needle dissection, the sperm are identified and separated from the seminiferous tubules and surrounding tissue. The majority of these sperm are immature, although some are motile or “twitching.” TESE samples contain a large amount of cellular debris. Finding sperm in the testicular tissue can be laborious and can take several hours to process depending on the degree of sperm production and the etiology of testicular failure. There are multiple processing methods that may be used to identify the sperm. Typically the testicular tissue is evaluated under a stereomicroscope to identify seminiferous tubules and remove blood clots. Following identification, the testicular sample is processed by dispersion of the tubules through mechanical mincing and/or enzymatic digestion. Once homogenized, the sample is evaluated using the inverted microscope (400× magnification) to identify the presence of sperm. The sperm are freed from the seminiferous tubules and other debris by dissection.
Fertilization
- ◆
Approximately 2 to 6 hours following oocyte retrieval, insemination is performed by conventional insemination (CI) or intracytoplasmic sperm injection (ICSI).
- ◆
In CI, sperm must traverse the layers of the oocyte, mimicking in vivo fertilization processes.
- ◆
ICSI requires the selection and immobilization of viable sperm, positioning of a denuded oocyte for injection, and rupture of the oolema before the sperm are released into the oocyte.
- ◆
Various techniques for sperm selection for ICSI exist and are described.
Fertilization is the process that results from the fusion of two parental gametes―the oocyte and the sperm. When these two gametes meet, a series of crucial events are set into motion that lead to fertilization and ultimately the development of a new individual (see Chapter 31 ).
In order for natural fertilization to occur, sperm must undergo capacitation to traverse the layers of the oocyte ( Figs. 32.3 and 32.7 ). Capacitation is a biochemical process involving hyaluronidase activity, which sperm use to navigate through the cumulus mass that surrounds the oocyte. The hyaluronidase enzyme is located on the outer membrane of the sperm head and is thought to be expressed when the spermatozoa are mature (see PICSI). To penetrate the ZP, spermatozoa bind and then undergo the acrosome reaction. The binding is mediated by sperm-specific receptors, primarily oocyte-specific ZP glycoprotein 3 (ZP3), which serves as both the primary ligand for sperm binding and a trigger for the acrosome reaction. The sperm then fuses with the oocyte membrane (oolemma) and gets incorporated into the oocyte via phagocytosis (see Fig. 32.7 ). Oocyte activation ensues and results in the decondensation of the sperm nucleus and completion of meiosis in the egg, with extrusion of the second polar body. Within 5 to 6 hours following sperm incorporation, new nuclear membranes form around the decondensed male and female chromatin, resulting in two pronuclei (2PN).
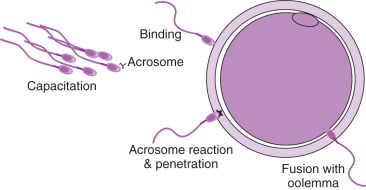
IVF is a process designed to join the egg and sperm outside the body. There are two methods for IVF that are performed in the laboratory: CI and ICSI. The indications are discussed in Chapter 31 . When performing CI, the sperm must traverse the layers of the oocyte more or less mimicking the natural process. However, with ICSI the sperm is directly injected into the oocyte, which circumvents the natural penetration of sperm through the cumulus, ZP, and oolemma. Once the sperm is injected, the natural events including oocyte activation must occur for normal fertilization to result. The oocyte activation requires a sperm factor (phospholipase C zeta) that causes the production of inositol-triphosphate (IP3) in the ooplasm and results in calcium release, which is required to complete fertilization. The process for each method of fertilization is described in the following text.
Conventional Insemination
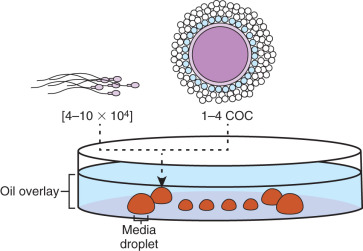
Insemination should be done within 2 to 6 hours after the egg retrieval. Evidence has shown that insemination prior to 2 hours postretrieval leads to a compromise in pregnancy rate. Significant delays after retrieval are associated with decreased fertilization and pregnancy outcomes due to spindle instability and subsequent loss or scattering of chromosomes in the oocyte. On average, 55% to 65% of the oocytes will normally be fertilized (2PN). The procedure involves using prepared dishes from the day prior to ensure proper equilibration of medium and oil (37°C and pH). Since incubation is required, insemination dishes are made of droplets composed of sodium bicarbonate–based media under an oil overlay ( Fig. 32.8 ). The droplets typically are 50 to 200 µL in size with one to four COCs in each. Oocyte grouping is determined by the laboratory and is mostly based on efficiency of the procedure. The cumulus is maintained on the oocytes to theoretically ensure maximum fertilization. The processed sperm are then added to the droplets. It is recommended to aseptically dispense 40,000 to 100,000/mL of motile sperm per microdroplet. Fertilization rates have been demonstrated to be significantly higher with 20,000 sperm per egg versus 5000 sperm per egg. This sperm concentration range is optimized for achieving high normal fertilization rates without the increased risk of polyspermy. It is critical that the insemination process be performed as quickly as possible or under an isolette with CO 2 and on a heated stage to reduce pH changes or cooling. Once insemination has been completed, the gametes are coincubated overnight, and 16 to 20 hours postinsemination, oocytes are observed to check for normal fertilization. In most instances, we like to do the insemination between 1 p . m . and 3 p . m . to optimize the timing for fertilization check the next day (see Fig. 32.1 ). Although traditionally inseminated oocytes are incubated overnight, sperm binding to the zona can take place within 1 to 3 hours after insemination and fertilization occur. Thus a few hours of sperm-oocyte contact can yield the same time course of events that was observed after overnight insemination. One thought is that longer exposure to the high concentrations of sperm may increase exposure of oocytes to high ROS levels and alter the pH in the insemination medium. These derangements may affect embryo viability, cause zona hardening, and lead to a lower pregnancy rate. Therefore some labs may choose to wash the excess sperm free from the oocyte after 3 hours incubation. Regardless of the incubation time, the current standard of care is to evaluate for fertilization 16 to 20 hours postinsemination ( Table 32.4 ).
| Technique | Requirements | Time Considerations | Advantage | Disadvantage |
|---|---|---|---|---|
| Intracytoplasmic sperm injection (ICSI) | Standard ICSI microscopes | Rapid selection of spermatozoon | Inexpensive solution for sperm selection | Selection of sperm is based on motility and morphology, does not exclude spermatozoa with DNA damage |
| Intracytoplasmic morphologically selected sperm injection (IMSI) | Microscope capable of 6000x magnification | Additional processing time required to search and select appropriate sperm | May increase implantation and pregnancy rates | Equipment cost can be prohibitive Sperm selection should take place at room temperature to prevent DNA damage |
| Hypo-osmotic swelling test (HOST) | Hypoosmotic solutions | Additional 30-120 minutes of processing prior to selection | Differentiates viable and nonviable sperm May be able to differentiate degree of DNA fragmentation | Over exposure to hypo-osmotic conditions can affect viability |
| Physiological intracytoplasmic sperm injection (PICSI) | Specialized dishes containing hyaluronan microdots | Additional 5-30 minutes of processing prior to selection | Assists in selecting mature sperm with normal morphology Selects sperm with increased DNA integrity May reduce rates of aneuploidy | Difficulty in micro-manipulating sperm without viscous media Relatively expensive |
| Sperm head birefringence | Microscopes equipped with contrast and polarizing lenses | Additional processing time required to search and select appropriate sperm | Selects mature sperm that have undergone the acrosome reaction Helps select sperm with lower DNA fragmentation and a higher degree of nuclear normalcy May improve Day 3 embryo quality and increase implantation rates | Equipment cost can be prohibitive |
However, with low sperm counts or poor motility, CI is associated with decreased fertilization. In some cases, improved fertilization may be achieved by increasing the number of motile sperm (see earlier discussion of semen preparation). In other instances fertilization can be improved by decreasing the volume of medium, which may increase the interaction between the gametes, such as the insemination technique involving a microdroplet under oil. This technique has been shown to raise the chances of fertilization to over 40% and to provide a clinical pregnancy rate of 24%.
Several methods have been developed to improve fertilization when the CI was poor or unsuccessful. These methods bypass layers of the COC to aid in fertilization. Initially, the cumulus was removed, but fertilization rates were not improved. The techniques subsequently focused on the ZP, given that it was a known barrier to sperm penetration. The various methods were intended to thin the zona so that the inseminated sperm could have direct access to the oolemma. The techniques included zona drilling (ZD), partial zona dissection (PZD), and localized laser cutting. However, the increased incidence of polyspermy and a still relatively low fertilization rate led to subzonal sperm injection (SUZI). This technique involves the direct injection of several motile sperm into the perivitelline space with an injection pipette. But even SUZI had its limitations, such as the inability to overcome acrosomal abnormalities or dysfunction of the sperm–oolemma fusion process. As a result, normal fertilization rates were poor (20%), and pregnancy rates were too low to consider PZD and/or SUZI for routine clinical application.
Intracytoplasmic Sperm Injection
ICSI is a procedure that entails microinjecting a single spermatozoon into the oocyte, bypassing the ZP and the oolemma. These micromanipulated oocytes undergo the normal process of egg activation, including Ca 2+ release. This suggests that contact of sperm and egg plasma membranes is not a critical step for egg activation. The first successful use of ICSI was to obtain live offspring in rabbits and cattle, and in 1992, the first human pregnancies and births were reported. ICSI was also found to be superior to SUZI with respect to fertilization rate, embryo yield, and embryo implantation rate.
The average fertilization rates with ICSI range from 70% to 80%, and pregnancy rates are comparable to those of cases with CI and normal sperm counts. Given the success, ICSI is now performed universally when CI cannot be performed because of poor sperm motility or morphology.
ICSI is performed on an inverted microscope equipped with micromanipulators and microinjectors under 200× and 400× magnification while on heated stage. The micromanipulators allow three-dimensional manipulation of coarse and fine movements. One microinjector is used to hold the denuded oocyte and the other to inject the sperm. Movement of the oocyte or sperm is achieved by a micrometer that controls a plunger. The whole setup is placed on a vibration-proof table to avoid possible environmental motions.
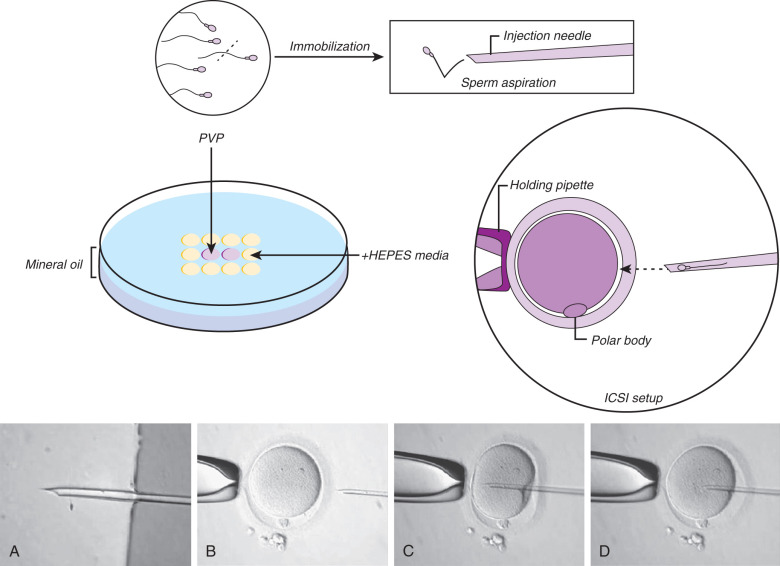
ICSI can be described with the following steps: (1) denudation of the oocyte, (2) selection and immobilization of a viable sperm, (3) positioning of the oocyte before injection, and (4) rupture of the oolemma before release of the sperm into the oocyte. Fig. 32.9 shows the injection sequence.
Denudation of the oocyte entails removal of the surrounding cumulus to visualize the oocyte and allow for a precise injection of the oocyte (also called oocyte stripping, Fig. 32.4 ). Since CCs are thought to secrete paracrine growth factors, which play a role in the nuclear and/or cytoplasmic maturation of the oocyte, several investigators have evaluated if early or delayed denudation will impact fertilization rates. The consensus suggests a preincubation time of 2 to 6 hours prior to denudation. However, some evidence suggests that there may be improvement in fertilization rates with longer preincubation times (>3 hours) and shorter times to fertilization once the oocyte has been denuded. Denuded oocytes are more vulnerable to pH changes.
Removal of the surrounding cells is typically achieved by a combination of enzymatic and mechanical procedures (see Fig. 32.4 ). Since hyaluronic acid is a major component of the extracellular matrix of the cumulus, these oocytes are repeatedly transferred to a hyaluronidase solution and HEPES buffered medium for approximately 30 seconds with pipettes of decreasing diameter (150 to 135 μm) to facilitate mechanical denudation. Hyaluronidase-purified preparations of bovine origin are most commonly used, although human recombinant hyaluronidase is available. Regardless of the preparation, hyaluronidase helps to digest the connections between the CCs, freeing the oocyte for grading and ICSI (see previous text). After denudation, oocytes should be thoroughly washed to remove traces of hyaluronidase. It should be noted that both the enzyme concentration and the duration of exposure to the enzyme should be limited because of the possibility of damage or parthenogenic activation of the oocytes.
Once denuded, the oocyte can be clearly visualized, and its morphology and maturation can be assessed (see Fig. 32.5 ). Oocytes that demonstrate a polar body (MII) and are morphologically intact are suitable for ICSI and can be fertilized accordingly. Oocytes that do not show the first polar body (MI) or that are GV can undergo extended culture and subsequently be micromanipulated if the polar body has been extruded. However, as previously mentioned, embryos derived from immature oocytes that mature in vitro after ovarian stimulation rarely result in pregnancies. Therefore GV oocytes are usually discarded. Denuded and rinsed oocytes are incubated until the time of microinjection. Oocytes should be fertilized within 6 hours after collection. However, some evidence suggests that shorter times to insemination once the oocytes have been denuded may improve outcome.
To inject a sperm into the oocyte, the sperm must be selected and immobilized. The process of selection starts with creating a plastic microinjection dish containing microdroplets of HEPES-buffered medium under a layer of mineral oil (see Fig. 32.9 ). Generally there is a central drop (~5 µL) of polyvinylpyrrolidone (PVP) surrounded by more microdroplets (culture medium to support the oocyte during ICSI). Because of the viscous nature of the PVP, the sperm’s progression is greatly slowed, and there is usually a general dispersion aiding in sperm selection and manipulation. The use of PVP also aids in microinjector use, as the viscosity allows for greater control of sperm with the injection needle. Once the semen has been processed, a small amount (~1 µL) of sperm suspension is added to the PVP drop and oocytes are added to the surrounding drops.
The injection pipette is backloaded with PVP, then a viable sperm is located. Viability is evidenced mostly by the sperm’s motility (sometimes only “twitching”) and morphology. Immobilization of the sperm involves rubbing, dragging, or knocking the sperm tail with the injection pipette against the bottom of the dish. This causes a breakage or rupture in the sperm tail’s plasma membrane. Aggressive breakage has been shown to increase fertilization rates. Immobilization of spermatozoa has been proven to be an important factor for oocyte activation, which is achieved by the release of sperm cytosolic factors via the ruptured membrane. Changes in the sperm plasma membrane and acrosome after immobilization have been confirmed by transmission electron microscopy and scanning electron microscopy. Mechanical immobilization with the ICSI needle is the general rule of thumb; however, use of a laser has been described, resulting in identical fertilization rates. After immobilization, the sperm cell is aspirated tail first with the injection pipette and moved to the microdrop containing the oocyte.
The oocyte is picked up and held in position with gentle suction by the holding pipette. The polar body of the oocyte should be at the 12 or 6 o’clock position, which may avoid possible damage to the meiotic spindle, although the first polar body does not always coincide with spindle localization. Nevertheless, there is little evidence demonstrating that spindle monitoring during ICSI reduces spindle damage or increases the chance of fertilization.
If both the holding pipette and the oocyte are in perfect focus, the injection needle, containing the immobilized sperm cell near the tip, can be introduced in the equatorial plane of the oocyte at the 3 o’clock position. Permanent focus of the injection pipette tip ensures that the needle remains in the equatorial plane of the oocyte. Passing through the ZP is fairly easy and is achieved by simply advancing the injection pipette. However, the oolemma is not always immediately pierced by simple injection of the needle. Minimal suction of the membrane into the needle or multiple passes of the needle into the oolemma may help breakage. Even selecting another area of the oocyte may be necessary. A sudden flow of cytoplasm into the needle or “pop” of the oolemma indicates membrane rupture. Once it is determined that the oolemma is broken, the sperm cell can be released into the oocyte with a minimal amount of surrounding media, and the needle then withdrawn.
Once ICSI is completed, all injected oocytes are rinsed and moved to culture microdroplets of bicarbonate-based medium under oil in conditions similar to the CI oocytes. These oocytes are then examined for integrity and fertilization approximately 16 to 20 hours after ICSI ( Table 32.5 ).
| Outcome | Example | CI (incidence) | CI (ploidy status) | T | ICSI (incidence) | ICSI (ploidy status) | T |
|---|---|---|---|---|---|---|---|
| 0 PN |  | 1% * | >50% diploid | Y | 1% * | >50% diploid | Y |
| 1 PN |  | 10% | 30–60% diploid | Y | 2–5% | Haploid | N |
| 2 PN |  | 55-65% | Diploid | Y | 70–80% | Diploid | Y |
| 3 PN |  | 0-6% | Dispermic triploidy † | N | 2–4% | Digynic triploidy † | N |
| >3 PN |  | Rare | Multiploidy | N | Rare | Multiploidy | N |
* incidence of 0PN that continue to cleave
Sperm Selection Techniques
Sperm selection varies based upon processing technique. For ICSI, the technician subjectively evaluates sperm at an optical magnification of approximately 200 to 400× for motility and morphology. However, spermatozoa appearing morphologically normal at this magnification may carry various structural abnormalities at the subcellular level; these remain undetected by the embryologist. Some evidence suggests that the fertilization rate, embryo development, and pregnancy outcomes can be improved with more elaborate selection.
Injection of Morphologically Selected Sperm
Bartoov and colleagues developed a method to evaluate sperm in real-time at high magnification called motile sperm organelle morphology examination (MSOME). MSOME is performed on an inverted microscope equipped with Normarski interference contrast optics, which allows observation at high magnification (<6000×). This method led to the development of the intracytoplasmic morphologically selected sperm injection (IMSI) procedure, which is based on sperm normality as defined by MSOME classification. The aim of this technology is to improve ICSI outcomes by selecting against sperm that have abnormalities, such as vacuoles, that are associated with DNA fragmentation.
The studies that compared IMSI with traditional ICSI show conflicting results. Several studies have demonstrated that IMSI significantly improves fertilization rates, embryo quality, blastocyst development, implantation, and pregnancy. In addition, IMSI has been shown to reduce miscarriage rates. However, in small prospective randomized trials with unselected infertile populations, IMSI did not provide significant improvements in clinical outcomes as compared with routine ICSI; however, severe male factor patients experienced significantly improved embryo development and implantation with IMSI. The most recent Cochrane review does not support the clinical use of IMSI because of lack of evidence-based benefit ; because of the added cost of equipment and the time-consuming nature of its use, large-scale prospective randomized trials are needed to determine the benefit of sperm selection using IMSI. For now, IMSI should be limited strictly to cases of severe male-factor infertility and repeated implantation failure following ICSI.
Hyaluronic Acid–Mediated Sperm Selection: Preselected or Physiologic Intracytoplasmic Sperm Injection
In contrast to visually selected spermatozoa, hyaluronic acid–mediated sperm selection is a technique that may reduce rates of fertilization with sperm of diminished maturity in ICSI. Hyaluronic acid (HA) is a high-molecular-weight glycosaminoglycan and is the main component of the extracellular matrix of the cumulus mass. PICSI uses a commercially available dish with microdots of hydrogel containing hyaluronan. Sperm is incubated on the dish for 30 minutes and those sperm that attach are then used for ICSI. Spermatozoa that are able to permanently bind to HA in vitro are mature and have completed the spermiogenetic process of plasma membrane remodeling, cytoplasmic extrusion, and nuclear maturity. Hyaluronan-bound (HB) sperm exhibit decreased levels of cytoplasmic inclusions, residual histones, and chromosomal aneuploidy and an increased expression of the heat shock–related 70 kDa protein 2 (HspA2) chaperone protein. Bound sperm have also been shown to have a high density of HA receptors. In contrast, immature spermatozoa with deficient plasma membrane remodeling are not able to bind to HA as there is a deficiency in the zona and HA binding sites. These unbound and immature sperm have a high retention of creatine kinase and are associated with meiotic defects as well as potentially higher rates of aneuploidy. Sperm bound to HA have also been shown to have lower levels of DNA fragmentation and have a higher percentage of nuclear normalcy compared with those selected by traditional ICSI techniques. Overall, HB sperm demonstrate increased developmental maturity and enhanced levels of functional competence over their nonbinding counterparts.
Several observational studies have demonstrated that the injection of HA sperm improves fertilization rates, embryo quality, and implantation capacity, while others found no differences in fertilization, pregnancy, and implantation rate. Most recently, a multicenter, prospective, randomized study demonstrated improvement in clinical pregnancy rates and a reduction in miscarriage with PICSI. Although there is no consensus on whether PICSI improves outcomes, it has not been shown to cause detrimental effects. More studies are needed prior to routine use (see Table 32.4 ).
Hypoosmotic Swell Test
In cases where there is an absence of native or stimulated sperm motility, assessment of viability is essential. The hypoosmotic swell test (HOST) is a simple viability test based on the semipermeability of the intact and physiologically functional plasma membrane. When sperm are exposed to hypoosmotic conditions, an influx of water to the sperm tail results in an expansion of the cell volume. Cells that are viable will respond to the osmotic pressure of the external solution by attempting to attain equilibrium with the solution. If the solution is hyperosmotic, the cell will shrink; if the solution is hypoosmotic, the cell will swell. When viable sperm are bathed in hypoosmotic solution, the tails coil and the heads swell within 10 seconds.
Nonviable sperm will not undergo these changes. Therefore this assay is effective in identifying viable nonmotile sperm for use with ICSI. Exposure of sperm for more than 2 to 5 minutes to a hypoosmotic solution may be damaging and should be avoided. Once a viable sperm is identified, the sperm must be washed two to three times in handling solution and then moved into the PVP solution before they are used for ICSI. Recently Stanger and colleagues showed a significant correlation between the HOST score and the degree of DNA fragmentation. They observed the lowest level of DNA fragmentation in the sperm’s presenting pattern or grade D or E tail (World Health Organization [WHO] criteria) folding during a HOST. Given the simplicity and low cost of this technique, investigators have proposed that this test alone or integrated with others may provide a method of standardizing the selection of sperm for ICSI.
Polarized Light Microscopy
Advances in microscopy have made the visualization of sperm heads’ birefringence feasible. Birefringence can be assessed with the addition of a polarization apparatus in an inverted light microscope used for ICSI. Evidence has shown there is a positive correlation between the sperm head’s birefringence and DNA fragmentation. Preliminary clinical studies have indicated that when sperm cells are selected based on their birefringence, a higher fertilization rate and better embryo development are demonstrated than with conventional ICSI. Another study showed that the implantation rate is higher (39.0%) with reactive sperm than with nonreactive sperm (8.6%). However, more studies are needed to determine the role for polarized light microscopy in the selection of sperm for ICSI.
Assessment of Fertilization
- ◆
Sixteen to 20 hours following insemination, fertilization is assessed by verification of pronuclear number and morphology.
- ◆
Abnormal pronuclear number is often associated with karyotypic abnormalities; however, it can also represent a deviation from the normal timeline of development.
- ◆
Multipronuclearity is often the result of polyspermy after CI, whereas triploidy may be the result of failed meiosis II (MII) with retention of a second polar body following ICSI.
- ◆
Total fertilization failure (TFF) results from abnormal sperm or inappropriate laboratory conditions.
- ◆
Rescue ICSI is associated with low pregnancy rates.
The fertilization rate is dependent on the denominator. Typically with CI, the denominator is all oocytes inseminated. The average fertilization rates for CI are 50% to 70%. The fertilization rate after ICSI is usually expressed per number of injected oocytes and ranges from 70% to 80%. Upon injection, oocyte damage (degeneration) can occur, with an incidence of 0% to 9%. The degeneration can be a result of the procedure, but most often it is due to postmaturity and is technician-independent.
The current standard of care is to evaluate for fertilization 16 to 20 hours post insemination. Fertilization assessment should be performed under high magnification (at least 200×), or a suitable time-lapse microscopy device, in order to verify PN number and morphology. Oocytes are considered normally fertilized when two individualized or fragmented polar bodies are present together with two clearly visible pronuclei (2PN) that contain nucleoli (see Table 32.5 ). These pronuclei contain one male and one female haploid chromosome complement. Typically, the two pronuclei develop simultaneously. However, a common finding seen after IVF is an abnormal pronuclear number (see Table 32.5 ). These embryos commonly have karyotypic abnormalities and in most cases are discarded. However, in some cases a portion of these embryos can represent a deviation from normal development. In other words, they can be diploid and produce viable pregnancies. In some cases, rather than discard these embryos, clinics may choose to extend culture while others will do preimplantation genetic screening (PGS) prior to transfer.
In the case of 0PN, the etiology is most often a failure of fertilization. The etiology can be due to failure of sperm penetration. However, if sperm has penetrated the oocyte or was injected, the etiology may be premature sperm chromosome condensation, failure of sperm head decondensation, or ejection of the sperm from the oocyte. The cause for sperm dysfunction can in part be attributed to oocyte immaturity. On the other hand, the absence of pronuclei could also mean early syngamy. Typically syngamy occurs 20 to 24 hours after insemination. Syngamy is a time period of pronuclei nuclear breakdown before proceeding through the first mitotic division to form the two-cell embryo. In one study of 22,308 fertilized oocytes assessed at 17 ± 1 hour postinsemination, 8% were already in syngamy. The significance of early syngamy is not clear. One study showed embryos with syngamy prior to 23 to 24 hours produce higher implantation rates, while another study showed that syngamy prior to 20 hours resulted in decreased live birth rate. Most clinics will recheck for early cleavage, and/or continue to culture these embryos and evaluate prior to transfer or do PGS.
1PN oocytes after CI are likely to be parthenogenetically activated as a result of mechanical or chemical factors. However, regardless of the fertilization method, the etiology of monopronuclear formation is largely unknown. In some cases, the zygote may be diploid and presence of 1PN may be explained by asynchronous pronuclear formation, or possibly male and female pronuclear fusion (atypical syngamy). Evidence suggests that more than half of unipronuclear embryos after CI are diploid, which is significantly higher than what is observed after ICSI (20% to 30%; Table 32.5 ). In vitro culture of these 1PN oocytes shows that although the majority are incapable of supporting early development, a small proportion exhibit further cleavage, are indistinguishable from 2PN embryos, and are capable of forming morphologically normal blastocysts resulting in live births.
Multipronuclear zygotes are characterized by the presence of more than two haploid chromosome complements (see Table 32.5 ). Triploidy (3PN) is the most common multi-pronuclear zygote. The extra pronuclei may be of maternal (dygynic) or paternal (diandric) origin. If CI, triploidy is most often due to insufficient protection against polyspermy (diandric). The etiology is not clear, but has been attributed to either a high sperm concentration or immature and/or postmature oocytes. However, with ICSI, the cause of triploidy is largely due to failure of meiosis II with retention of the second polar body (dygynic). Few studies have suggested that retention of the second polar body after ICSI could be induced by a deterioration of the meiotic spindle during sperm injection.
Triploidy may also be related to oocyte competence or sperm quality. Several studies have shown that 3PN is related to ovarian stimulation and/or increased maternal age. Others have shown that the presence of 3PN after ICSI is associated with a global impact on the remaining cohort of embryos, resulting in worse clinical outcomes. On occasion, especially in cases with male infertility, diploid sperm (failure of errors in male meiosis I and/or II) can be the etiology. The frequency of diploid spermatozoa in infertile, particularly oligozoospermic, men may amount to ≤1.9%, compared with 0.2% to 0.3% in controls. Diploid spermatozoa account for up to 33% of triploidy after ICSI. It is also possible that triploid embryos could result from fertilization of a diploid oocyte (i.e., giant oocyte) by a single sperm (digynic). Triploidy occurs in up to 6% after IVF and 15% to 18% of cytogenetically abnormal cases after genetic testing of the products of conception in the setting of spontaneous abortions. Finally, these triploid zygotes can result in pregnancies with severe congenital anomalies and should not be transferred.
More than 3PN has also been observed. Tetraploidy can be the result of a diploid sperm injection and retention of the second polar body. On the other hand, there is also evidence for polyspermic fertilization of a normal oocyte by three independent normal sperm. In CI cases, the etiology has been attributed to failure of oocyte to block polyspermy or high sperm concentration. Another possibility is the confusion of pronuclei with cytoplasmic vacuoles. These zygotes should not be transferred.
Failed Fertilization Options
TFF usually refers to the failure of all available mature MII oocytes to fertilize, and the etiology may be distinctly different than with low fertilization. The incidence of TFF after CI is 5% to 10% and after ICSI is 2% to 3%. The etiology of TFF after CI is mostly unknown and often is not elucidated, but is thought to be either due to sperm abnormalities (as with low fertilization) or a result of “shock” due to laboratory conditions. Sperm abnormalities are more likely the cause when there is a total absence of sperm binding to the ZP or failure of the sperm to successfully penetrate the ZP. Since the recurrence rate of TFF or low fertilization rate with CI is 30% to 70%, the most common treatment for failed/low fertilization after CI is ICSI the following cycle. Although ICSI may be performed emergently to salvage fertilization, successful pregnancies with this technique are rare (see the discussion of rescue ICSI ). The etiology of TFF after ICSI is more likely due to failure of oocyte activation, which can be attributed to either gamete. After TFF with ICSI, low or moderate fertilization is observed in 30% of repeated ICSI cycles. If there is failed or low fertilization after ICSI, the next step is to try to improve on selection of sperm (see previous text) or use oocyte activation procedures (see subsequent text).
Rescue Intracytoplasmic Sperm Injection
Rescue ICSI is an option after failed fertilization with CI that involves the microinjection of unfertilized oocytes with a single sperm from the original sample. This can take place the following day (late rescue) or 8 to 10 hours after oocyte retrieval (early rescue). In some countries this procedure is banned (e.g., the United Kingdom) as it cannot be determined if fertilization has occurred and is just delayed. Others reserve this procedure for complete fertilization failure following CI. Fertilization rates from rescue ICSI have been shown to be low, and the generated embryos achieve lower pregnancy rates. Although controversial, the poor outcomes have been attributed to chromosomal abnormalities and/or dysynchronous embryo development with respect to the endometrium. The higher aneuploidy might be the cause of the failed fertilization or the result of the in vitro aging process. Preliminary evidence suggests that pregnancy rates can be improved with the performance of early rescue ICSI or frozen embryo transfers. Some centers do perform rescue ICSI, but because of the low pregnancy rate and the potential genetic risk, more studies are needed before rescue ICSI can be considered a standard of care.
Artificial Oocyte Activation
Artificial oocyte activation has been proposed as a way to overcome TFF after ICSI. Calcium ionophores, such as ionomycin or calcimycin (A23187), are the most popular artificial activating substances for human oocytes. The procedure entails first microinjecting the sperm into the oocyte, followed by exposing the oocyte to an ionophore solution on two separate occasions. However, there is no standardization of the procedure as there are disparities in type, concentration, exposure time, and, mode of application of the ionophore among published reports. The lack of standardization may contribute to the discrepancies in outcomes in the literature. Evidence has shown that the success of ionophore treatment is related to the fertilization rate in a previous cycle. The cutoff value for fertilization rate in the preceding cycle that distinguished between an increase in fertilization rate in the presence of A23187 and no improvement was 30%. Although studies have shown a significant increase in fertilization rates with artificial oocyte activation (up to 70% to 77%) and developmental outcome of children aged 3 to 10 years are within expected ranges, the numbers of live births registered are low and the lack of long-term follow-up after after ionophore treatment does not allow proper calculation of the actual risk. Therefore this treatment remains experimental.
Grading
- ◆
Time-lapse imaging (TLI) suggests that the timing of embryonic cleavage events may be associated with embryonic developmental competence.
- ◆
Day 3 embryo grading entails the assessment of blastomere number as well as degree of symmetry and fragmentation.
- ◆
Embryos may be cultured to the blastocyst stage for selection or for PGS. Blastocyst scoring systems evaluate the degree of expansion in addition to the quality of the inner cell mass (ICM) and trophectoderm (TE).
Fertilized oocytes remain in incubators until specific, clinic-dependent time points are reached, at which time development is assessed by embryo grading and embryos are transferred or cryopreserved. Embryos are graded to determine viability and in order to select embryos for transfer. Given that embryo development follows a programmed time line, assessing embryos for viability should be standardized along a set of specific time points. The oldest and most common method of embryo grading is based on morphologic measurements (see further on). Some clinics assess the embryos only once prior to transfer, biopsy, or cryopreservation while others assess every day.
Although more morphologic assessments may improve embryo selection, the majority of laboratories limit the number of observations due to the potential stress of removal from the incubator on the embryo. Removing the embryos from a controlled environment may result in temperature and pH fluctuations that might lead to significant stress. The introduction of time-lapse incubators has the potential to overcome this dilemma. Time-lapse incubation (TLI) is a closed culture system that allows continuous monitoring and observation of exact time points of cell division via TLI without the need to remove the embryos from the incubator. Therefore TLI allows virtually unlimited morphologic assessments and observation of cellular events that would otherwise remain undetected by conventional methods such as development kinetics (duration of embryonic events), cleavage anomalies (i.e., one- to three-cell divisions, reverse cleavage), and fragment reabsorption. Indeed, evidence has shown that cleavage anomalies that would not be seen with conventional grading and early developmental kinetics such as the timing of pronuclear breakdown (PNB) are associated with aneuploidy. Several early studies have suggested with bioinformatics prediction models can predict blast development, embryo quality, and live birth. However, these prediction models could not be widely reproduced in other clinics. Therefore the expense of time-lapse incubators and concerns regarding implications for embryologists’ productivity in the face of unclear benefit for pregnancy outcomes has limited their use.
More studies are needed before TLI is widely accepted as the routine culture system. It is possible that with more validation and increasing numbers of recordings on the timing of developmental behaviors that we will see improvement in clinical outcomes with TLI. The convenience of grading at any time, gaining additional morphokinetic information, and a closed-system approach is attractive. However, until TLI is either cost-effective or studies show clinical improvement with its use, grading embryos at key time points by way of stereoscopic microscope observation prior to transfer will be the preferred method of assessment by most laboratories.
Embryo Development and Selection
As early as the pronuclear stage (16 to 18 hours), morphologic scoring, including alignment and size of pronuclei as well as alignment and number of nucleoli, can be performed. While some studies have shown a prognostic effect of PN scoring for pregnancy and aneuploidy, others have not been able to demonstrate the association. However, TLI has shown that the timing of PNB is associated with aneuploidy and implantation and is a key assessment for embryo selection. Of course, the timing of assessment is critical because pronuclear development is a dynamic process; therefore, zygote scoring and/or the timing of PNB should be used with caution and only in conjunction with other methods of evaluation.
Postfertilization, approximately 90% of 2PN oocytes cleave, resulting in multicellular embryos. Studies have shown early cleavage to be a strong biologic indicator of embryonic potential and may be used as an additional criterion for embryo selection. Cleavage is conventionally assessed approximately 25 to 27 hours after fertilization. TLI has also shown that the duration of first cytokinesis may be associated with development outcomes. The early cleavage may be a sign of molecular health and cytoplasmic maturation. Previous studies have found that embryos with early cleavage have higher blastocyst development rate and are associated with higher pregnancy rates as compared to late cleavage embryos; however, other studies have not confirmed these findings. Interestingly, very early cleavage (<20 hours postfertilization) is associated with poor clinical outcomes. When using early cleavage as a predictor for embryo implantation, it may be necessary to consider the potential confounding factors that influence cleavage, such as culture conditions, stimulation protocols, and fertilization methods.
It is generally accepted that a day-3 embryo represents a key time point for embryo development. At this juncture, it is possible to identify embryos with better developmental potential. To obtain this stage of development, there is a required transition from relying on the maternal genome to the activation of the embryonic genome. Therefore, until recently, most centers cultured embryos for 3 days prior to embryo transfer. The embryo(s) transferred would then be selected based on morphology.
There are several grading schemes for cleavage stage embryos, all designed to determine likelihood of implantation. The Society for Assisted Reproductive Technology (SART) grading system approach was developed to help laboratories establish a common standard for embryo grading ( Table 32.6 ). Regardless of the system, the main parameters include the number of blastomeres, symmetry, and fragmentation. In addition, most laboratories evaluate for compaction and note if multinucleation is present.
| Cleavage Stage: Cell Number (1 to >8) | Morula/Blastocyst: Early/Expanded/Hatching | |||
|---|---|---|---|---|
| Grade | Fragmentation (%) | Symmetry | Inner Cell Mass | Trophectoderm |
| Good | 0 1%–10% | Perfect | Good | Good |
| Fair | 11%–25% | Moderate asymmetry | Fair | Fair |
| Poor | >25% | Severe asymmetry | Poor | Poor |
The single most important indicator of embryo viability at the cleavage stage is the occurrence of cellular division. The frequency of mitotic divisions is related to developmental potential of the embryo. Numerous studies have shown that the number of blastomeres is strongly associated with chromosomal abnormalities, implantation, and pregnancy outcome. Normally developing, good-quality embryos should reach the four-cell and eight-cell stages, respectively, on day 2 (40 to 44 hours) and on the morning of day 3 (64 to 68 hours) postmicroinjection ( Fig. 32.10 ). Studies have shown that embryos with either lower or higher cell numbers have significantly reduced developmental potential. Day-2 embryos with four cells result in a significantly higher implantation rate than 2- or 3-cell embryos. When transferring on day 3, embryos that comprise seven and eight cells have lower rates of chromosomal abnormalities and higher developmental potential than embryos with fewer or even higher numbers of blastomeres.
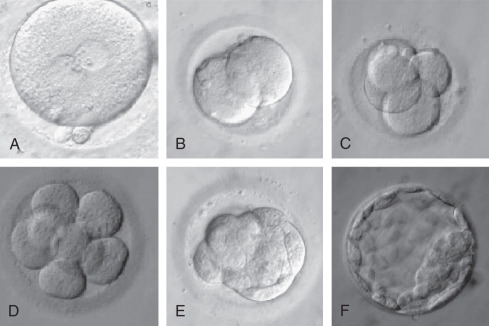
TLI studies have shown that the duration of cleavage events as well as the time to specific embryonic stages are associated with developmental potential. Although some studies suggest that early kinetics is most predictive of subsequent embryo development, others suggest that later kinetics are most predictive of blastocyst development. These studies suggest that optimal cleavage times fall into the two middle quartiles and embryos that reach the eight-cell stage fastest are associated with higher implant potential. Although morphokinetics are indicators of blastocyst development, there is low sensitivity and a lack of evidence to predict implantation above and beyond static morphologic assessments. It is likely that morphokinetics are affected by ovarian stimulation protocols, culture conditions, fertilization method, and individual differences.
Blastomeres should cleave synchronously and give rise to two cells with half the volume of the parent cell. Several studies have shown that even numbered embryos have higher implantation potential. In addition, a number of TLI studies have shown that abnormal early cleavage events—such as direct cleavage, where embryos cleave from two cell to three cells or exist as two cells for a limited time (<5 hours) or have uneven blastomere cleavage (>25% difference)—are associated with poor implantation potential. Examples of variations in blastomere size and cell generations are shown in Table 32.7 . Because embryos with uneven cell cleavage have a lower developmental capacity compared with evenly cleaved embryos, there is an emphasis on blastomere size within conventional embryo scoring systems. Therefore embryos are typically scored according to equality of size of the blastomeres in addition to number.




Numerous studies have shown that the presence of anucleate material or fragmentation is negatively associated with decreased blast development and pregnancy outcomes. Fragmentation grading is shown in Table 32.7 . Fragments form when segments of the cytoplasm protrude from the cell upon cell division and then separate. Blastomere fragmentation is observed in approximately 40% of early human embryos. The cause of fragmentation is not completely known. However, several theories include insufficient adenosine triphosphate (ATP) production, apoptosis with subsequent loss of regulatory proteins, and poor-quality gametes. Although evidence suggests that fragmentation may occur in vivo, in vitro conditions such as a decrease in pH, high oxygen tension, or decreases in temperature may lead to destabilization or loss of integrity of membranes and result in fragmentation. Studies have shown a trend toward increased fragmentation percentage with aneuploidy; however, there is no correlation with maternal age. Although several studies have shown that increased fragmentation percentage is associated with decreased implantation rate, the correlation of fragmentation score with outcomes remains controversial, as even highly fragmented embryos result in live births. This may be due to fragments being associated with mosaicism rather than aneuploidy and embryonic self-correction (see the discussion of preimplantation genetic testing [ PGT ]). In summary, fragmentation is a predictor of implantation but should not be the only morphologic criterion assessed.
Multinucleation is a frequently observed phenomenon in cleavage-stage embryos ( Fig. 32.11 ). Evidence has shown that multinucleated cells occur in cleavage stage embryos at the two- to four-cell range in 17% of cases and up to 65% in the third cleavage division. The etiology of multinucleation within a cell involves errors in cytokinesis. Multinucleation affects morphokinetic parameters. The presence of one or more multinucleated blastomeres (≤50%) has been associated with impaired cleavage and increased fragmentation, a poor prognosis for blastocyst formation, and reductions in the ongoing implantation rate, probably due to chromosomal abnormalities. The proportion of multinucleated blastomeres within one embryo is an important parameter. However, not all multinucleated embryos are chromosomally abnormal. Excluding these embryos for transfer when other mononucleated embryos are available is prudent practice.
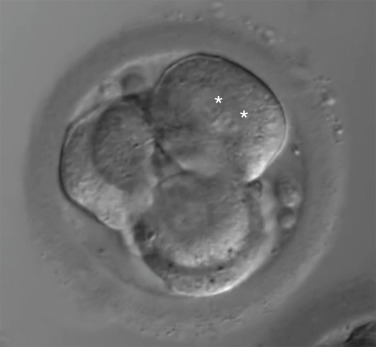
Assessing the degree of compaction is valuable because early compaction on day 3 may be associated with increased implantation potential. On day 4 (sometimes on day 3), a certain degree of compaction can be observed. Compaction is a morphologic change in a preimplantation embryo that results in a mass of cells held by tight junctions and is a fundamental event that leads to the formation of the TE, the ICM, and the blastocoele. Full compaction (16- to 32-cell stage) is followed by immediate cavitation and blastocoele expansion.
In the past it was suggested that the culturing of embryos to the blastocyst stage could naturally select the most viable embryos, increase implantation, and decrease miscarriage rates following a transfer. However, at that time it was difficult to maintain embryo viability to the blastocyst stage. With advancements in commercially available culture media, the common practice at present is to extend embryo culture for 5, 6, and 7 days to the blastocyst stage. Culture systems for blastocyst development may entail refreshing the media on day 3 and possibly on day 5 of culture with similar media, or sequential media systems may be used where media with different constituents specific to the phase of embryonic development are utilized. Other culture systems use one medium without refreshing. The specific culture system used for blastocyst development is dependent on laboratory conditions, clinic success rates, and preference. On average, the blastocyst rate is 50% but is dependent on culture environment and patient characteristics, such as age. As a result of the success with extended culture, transfers with blastocysts are commonly performed.
With improved selection, a blastocyst transfer would allow the transfer of fewer embryos, thereby decreasing the number of multiple pregnancies. However, although blastocyst culture improves selection and pregnancy rates are increased per transfer, the superiority of blastocyst transfer over early embryo transfer per cycle remains to be proven. It is essential for an embryo to develop into a blastocyst to implant, but not all blastocysts will implant and some viable embryos may fail to develop into a blastocyst in vitro. Extended culture also allows for preimplantation genetic screening (see the discussion of PGT ), although the safety of extended culture is not known. It is possible that prolonged culture to the blastocyst stage may increase the potential of aberrant epigenetic programming events. The decision to culture embryos to the blastocyst stage is based on the clinical scenario (see Chapter 31 ) and the laboratory’s capabilities.
Several scoring systems have been created for the evaluation of blastocysts. The most common classification system is shown in Table 32.8 . A distinction between early and expanded blastocysts is made, and the latter category is further scored according to the quality of the ICM and the TE. Ideally, blastocysts should have a thin ZP, smooth cohesive trophoectoderm, equality and close adhesion of blastomeres, a clearly visible blastocele cavity, and well-developed ICM with many closely aggregated cells. Evidence has shown that the single most important scoring factor in determining the likelihood of implantation is the TE score. Although top-quality blastocysts are associated with increased implantation potential, the correlation between blastocyst score and aneuploidy is modest, as more than 50% of aneuploid embryos are capable of achieving good- or excellent-quality blasts.







Improving Implantation Rates
- ◆
Assisted hatching involves the disruption of the ZP of cleavage-stage embryos and has been associated with increased implantation rates in some populations.
- ◆
PGS allows for selection of a euploid embryo for transfer, thereby increasing pregnancy rates on a per transfer basis. (Although PGS was initially performed on cleavage-stage embryos using fluorescence in situ hybridization [FISH] without benefit, advances in genetic testing platforms have yielded increased accuracy.)
- ◆
PGS will not increase the cumulative live birth rate per cycle, and whether it decreases the time to live birth remains to be determined.
It is generally accepted that human embryos generated by IVF have a low implantation potential: a 10% to 15% rate of implantation per embryo is routinely reported in the literature. Some have suggested that premature transfer of the embryo into the uterus may account for these low implantation rates. Data on the transfer of human cavitating morulae or blastocysts on day 4 or 5 of development indicate that such embryos have a higher implantation rate. In the case of blastocysts, implantation rates that are twice those of cleavage-stage embryos have been reported. However, many cleavage-stage embryos do not reach the blastocyst stage in vitro, and it is not clear how many of these embryos would have implanted had they been transferred at the cleavage stage.
Consequently, a remaining challenge in assisted reproduction is to overcome the obstacle of low implantation rates. Several factors, apart from synchronization between the endometrium and replaced embryos, are responsible. On one hand, endometrial receptivity is a factor in successful embryo implantation. On the other hand, embryonic factors play a major role in the implantation process. First, implantation failure may result from developmental arrest due to aneuploidy observed in human preimplantation embryos. PGS programs have been proposed to address this issue (described in more detail later). Second, to implant, the preimplantation embryo must escape from its ZP by means of a process called hatching. Following fertilization, the main function of the zona is protection of the embryo and the maintenance of its integrity.
Assisted Hatching
Developing embryos show a gradual thinning of the ZP during culture. The mammalian late blastocyst both expands and thins its ZP through alternating expansion and contraction. Intrinsic processes allowing thinning before successful hatching include pressure of the blastocyst as it expands against the zona and a lytic mechanism mediated by zona lysins. Finally, the ZP ruptures, allowing the embryo to hatch and implant into the endometrium.
An important observation leading to the clinical introduction of assisted hatching was the finding that there were higher implantation rates from embryos that were fertilized using microsurgical techniques. In addition, there was some evidence that cleaved embryos with thinner zonae had higher implantation rates than those with thick ZPs. Many embryos may not succeed in hatching from the zona for different reasons. A naturally thick ZP or hardening of the ZP due to prolonged in vitro culture conditions may impair or even prevent the natural hatching process, leading to implantation failure. Suboptimal culture conditions may cause TE lysin production to fall below the threshold level needed to promote zona thinning before successful hatching and subsequent implantation. Alternatively, failure of intrinsic lysin production unrelated to culture conditions may inhibit normal hatching.
To overcome hatching failure, three different micromanipulation procedures (mechanical, chemical, and laser-induced hatching) have been used to produce holes in the ZP of cleavage-stage embryos or to thin the ZP superficially. The advantages and disadvantages are shown in Table 32.9 and the methods are described in the following paragraphs.
| Hatching Methods | Advantages | Disadvantages |
|---|---|---|
| Mechanical | No effects of chemicals or heat during the drilling procedure Has not been shown to negatively affect fertilization or embryo development Inexpensive | Requires a double tool holder Time-consuming Requires high technical skill PZD openings can be narrow and can induce more trauma to blastomeres during biopsy and may result in lower hatching rates |
| Chemical | The size of the hole in the zona pellucida can be increased, which may enhance hatching Relatively inexpensive | Requires a double tool holder Difficult to regulate hole size Acid tyrodes can be toxic and can change intracellular pH leading to cytoplasmic degeneration |
| Laser | No need for disposable micropipettes Consistent results and hole sizes Lower degree of blastomere lysis compared with acid tyrodes Fast and easy to perform | Equipment cost can be prohibitive Laser drilling creates an aperture in the shape of a groove, which can cause constriction of the blastomere and result in lysis |
PZD is a form of mechanical hatching that was developed to create an artificial opening of the ZP of early cleavage-stage embryos. This technique is performed by stabilizing the embryo with a holding pipette while the zona is pierced with a microneedle pushed tangentially through the space between the ZP and blastomeres until it pierces the zona again. This spearing leaves a small part of the zona trapped against the microneedle. Rubbing this part of the zona against a holding pipette leaves a narrow incision in the zona. The mechanism of PZD is quick to perform; however, it produces holes of variable size that may not always be optimal. A modification of PZD has been described where a second cut is made in the zona below the first cut at a right angle. This leaves a cross-shaped hole on the surface of the zona pellucida (3D-PZD). This technique allows for the creation of a larger opening while maintaining the protection of the zona flaps during embryo transfer.
Chemically assisted hatching involves the use of acidic Tyrode solution (pH 2.35) to “drill” the ZP. A microneedle filled with acidic Tyrode solution is used to produce an opening at the 3 o’clock position, facing either empty perivitelline space or extracellular anucleate fragments. The acidic solution is expelled gently over a small area (30 µm), with the tip of the needle held very close to the zona until an opening is obtained. The mean diameter of the gap created is 20 ± 6.7 µm (range, 10 to 36 µm). It should be noted that the acidic Tyrode solution is embryotoxic and may alter the viability of the embryos. Therefore, with the advancement of alternative methods, chemically assisted hatching is rarely used today.
Laser-assisted ZP drilling has been shown to be a suitable alternative to the use of acidic Tyrode solution for zona dissection. Several types of laser sources have been investigated. In a convenient setup, the laser beam is guided through an optical lens and focused on the biologic specimen. There is no need for holding or cutting tools. Lasers with different wavelengths have been proposed to operate in this no-touch mode; however, infrared radiation is the most appropriate, especially when mutagenic risks are taken into account. The 1.48-µm diode laser allows rapid, precise, and easily controlled lysis of the ZP. The size of the hole can be chosen by varying the irradiation time; typically, a hole with a diameter of 20 µm is produced within 12 to 30 ms irradiation time. Larger-diameter holes are obtained by increasing the irradiation time.
Piezo technology, an alternative to laser technology, also produces a precise and controllable defect in the zona without embryotoxic chemicals. The vibration produced by the piezoelectric pulse is used for the assisted hatching. The ZP is carved easily in a limited area that is conical and approximately 30 µm in diameter. Five to eight applications in adjacent areas are needed to treat approximately one-third of the visible circumference of the ZP. Thereafter, weaker vibrations are used to create an opening 20 µm in diameter in the thinned zona at the junction between blastomeres.
Assisted hatching techniques designed to facilitate embryonic escape from the zona have been used in IVF centers since 1992. The initial indications for assisted hatching were patient age, zona thickness, high basal FSH value, and repeated IVF failure. Several studies assessing assisted hatching in these cases have given disparate results. For example, there remains conflicting evidence of a benefit of assisted hatching in patients with advanced maternal age. Some of the controversy may be due to the different methods used for assisted hatching and the lack of reproducibility of the hole sizes obtained. Disparate patient selection, retrospective study designs, and the lack of an appropriate control group may also explain conflicting findings in previous research. A recent meta-analysis that included randomized controlled trials of assisted hatching versus no assisted hatching and reported live births, clinical pregnancies, or implantation rates, concluded that there is some evidence that assisted hatching may increase the chance of pregnancy in women for whom IVF has been repeatedly unsuccessful and in older women, although definitive recommendations could not be given. As a result, the clinical relevance of assisted hatching of cleavage-stage embryos remains debated.
Preimplantation Genetic Testing (PGT)
For the past 25 years, PGT has been used to select against embryos carrying various forms of genetic abnormalities ( Fig. 32.12 ). There are two types of PGT: preimplantation genetic diagnosis (PGD) and PGS. Although the usefulness of PGD is definitive, the utility of PGS remains an area of active investigation.
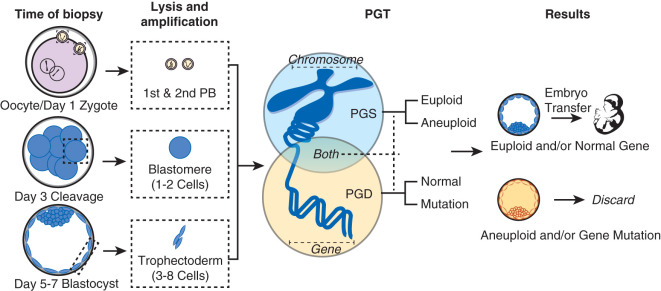
In 1989, PGD was introduced as a way of identifying genetic mutations in human embryos during an IVF cycle. Today, PGD is used for single-gene disorders, for inherited chromosome abnormalities, or human leukocyte antigen (HLA) matching. The aim of PGD is to avoid the transmission of genetic conditions in the offspring of a couple with increased risk of transmitting a genetic pathology, allowing the transfer of genetically healthy embryos. In principle, the indications for PGD are similar to those for prenatal diagnosis after chorionic villus sampling or amniocentesis, given that PGD at the single-cell level is technically possible.
In 1993, PGS was introduced based on the hypothesis that selection of euploid oocytes and embryos during assisted reproduction would lead to increased implantation rates and reduce spontaneous miscarriage rates. The hypothesis is based on the findings that show that the majority of implantation failure has been attributed to chromosomal aneuploidies. As previously mentioned, grading of the embryos and extended culture has, at most, a modest correlation with chromosome copy number. With the additional genetic testing, the aim is to increase the chance of delivery by preselecting euploid embryos for transfer. The number of referrals for PGS has only increased since its first clinical application, but the benefits of PGS remain a topic of debate.
An informed discussion of the utilization, successes, and challenges of PGT is predicated on a detailed understanding of how genetic material is obtained from the embryo. In this section, we describe the methods for each type of biopsy and discuss the advantages and disadvantages of each. The embryo biopsy procedure, regardless of stage of development, always involves two steps: opening the ZP and removing the cellular material. The methods for opening the ZP are similar to assisted hatching (see Table 32.9 ). However, the biopsy process for each developmental stage of the embryo is different. Three different sources of genetic material are potentially available for PGT: polar bodies from the initial conception, blastomeres from early cleaving embryos, and TE cells from embryos reaching the blastocyst stage (see Fig. 32.12 ). Each of these three types of biopsies is described in further detail in the following text. Next, we discuss the strengths and limitations of various modalities for genetic testing of biopsy samples. Lastly, we will end this section with a discussion of the clinical utility of PGD and PGS.
Timing of Biopsy
Polar-Body Biopsy
Polar-body biopsy is an option if only maternal mutations are investigated or as a method to avoid invasive embryo analysis. Polar bodies are byproducts of meiosis and have no biologic role in fertilization or in embryo development. Therefore, removal of either the first and/or second polar body for the purpose of genetic testing should not have a deleterious effect on a developing embryo. Genetic analysis of the first polar body was first performed in 1990 by Verlinsky and colleagues and has since been used to successfully screen for maternal mutations in cases of many single-gene defects. The polar-body biopsy is appealing for couples with ethical concerns as a preconception genetic tool and in countries (e.g., Switzerland, Germany) where genetic testing on cells derived from preimplantation embryos is prohibited. In such countries, polar-body biopsy may be the only method of tissue sampling for PGS. The merits of polar body biopsy for PGS are discussed later in this section.
In focusing on the general and practical limitations of polar-body biopsy, one must understand that it is labor-intensive. This is because more biopsies are performed per case, as (1) both polar bodies may need to be biopsied to increase accuracy and (2) only a portion of the prezygotes that are biopsied will fertilize and/or develop into embryos to be transferred. Other limitations of polar-body biopsy include that the polar body is prone to fragmentation and only a small amount of genetic material is available for analysis. Additionally, no information is gained regarding abnormalities of paternal origin, embryo sex, or subsequent mitotic errors.
Polar-body biopsy can be very useful for PGD. However, there are several challenges in doing a polar body biopsy for PGD. Because an oocyte has either a normal or abnormal copy of a gene, biopsy of the first polar body allows for carrier screening for genetic diseases, as the status of the PB will indirectly determine the status of the oocyte. For single-gene disorders, occasional recombination of homologous chromosomes requires information on both polar bodies. Therefore two separate biopsies and analyses are required, which doubles the cost of the analysis. Due to limited DNA availability, the detection of genetic disorders requires amplification of the DNA by polymerase chain reaction (PCR). A major concern in single-cell PCR is that it is very sensitive to contamination and susceptible to failure of amplification, leading a misdiagnosis or allele drop out (ADO). However, high accuracy in PGD for single-gene disorders can be achieved by application of sequential analysis of the first and second polar bodies in combination with multiplex PCR to detect allele dropout and contamination.
Polar-body biopsies would, in theory, be ideal for PGS if most embryonic aneuploidy were derived from nondisjunction in MI (as classically described). However, new findings suggest that less than half of the aneuploidy occurs in MI. Therefore a challenge associated with polar body chromosome analysis is related to making the decision to discard oocytes with aneuploid polar bodies or based on the aneuploidy of the first polar body. Evidence has shown that 4% of aneuploid polar bodies came from an oocyte which subsequently yielded an euploid zygote, while others have reported the birth of a chromosomally normal child resulting from an oocyte with reciprocal aneuploid polar bodies. As a result, both first and second polar bodies must be obtained for PGS.
In addition, reports from the European Society of Human Reproduction and Embryology (ESHRE) PGS Task Force pilot study, as well as others, reveal a high incidence of meiotic errors found in oocytes due to premature separation of sister chromatids (PSSC). PSSC is distinctly different than nondisjunction in that single sister chromatids prematurely segregate to the metaphase II oocyte but then subsequently segregate randomly in MII. As a result, about half of all errors in the first meiotic division (first polar body) are balanced in the second meiotic division (“self-correct”) with the chromatid segregating to either the second polar body or the zygote. In nondisjunction, the retention of both sister chromatids in MI yields as abnormal embryo. Since PSSC represents the majority of errors in meiosis I, the resulting PGS of the first polar body is difficult to interpret.
Currently, there is no consensus on the effectiveness of PGS with polar-body biopsies. The ESHRE PGS Task Force has launched a multicenter randomized controlled trial (ESTEEM trial) to determine the utility of polar-body PGS. ESTEEM is a randomized study of 560 cycles to evaluate PGS with comprehensive chromosome screening (CCS; see further on) using polar body biopsies in women between 36 and 41 years of age.
Polar-Body Biopsy Procedure
The first polar body can be removed from the oocyte on the day of oocyte collection between 36 and 42 hours after injection of hCG as long as the oocyte has extruded the first polar body. The second polar body can be removed from the zygote between 18 and 22 hours postinsemination. It is not recommended that both the first and second polar bodies be removed simultaneously as the first polar body may have degenerated by this time, leading to diagnostic error. There are several ways to biopsy polar bodies. In the past, the most common method was to breach the zona by mechanical means. A beveled pipette (12 to 15 µm in diameter) can be used for mechanical perforation of the ZP; once inside the perivitelline space, the polar body can be drawn out by aspiration into the pipette. If the polar body is still attached to the ooplasm, additional incubation may be required to permit complete extrusion. In addition, the use of a laser to breach the zona for biopsy is safe and yields equivalent blastocyst development to unbiopsied oocytes. Montag and colleagues also demonstrated equivalent results in comparing laser and standard ZD. The use of acid Tyrode solution for polar-body biopsy should be avoided. Studies using human oocytes show that, despite fertilization, Tyrode solution has an inhibitory effect on embryo development. This is thought to be primarily due to the direct effect of acid on the oocyte spindle.
Cleavage-Stage Biopsy
Cleavage-stage biopsy occurs on day 3 of development. At this stage, all of the cells are still totipotent (as confirmed by lineage tracing) and the embryos are not yet compacting. However, cellular totipotency wanes with further embryonic development and may not be an all-or-none property (i.e., sister blastomeres could express it to different degrees). Blastomeres of cleaving mammalian embryos can replace the loss of one or two blastomeres given that individual blastomeres are allocated but not committed to specific pathways. Apart from the reduction in cellular mass, the development of human embryos to the blastocyst stage in vitro is unaffected by biopsy and removal of one cell at the eight-cell stage. This means that up to one-eighth of the embryo can be removed without impairing its further in vitro development. Similar results were obtained in a mouse model.
When Handyside and colleagues reported the first pregnancies from the clinical application of PGD, it became clear that in vivo development was not impaired by the biopsy procedure. In 1994, 32 pregnancies worldwide were reported after PGD using cleavage-stage biopsy, with 29 infants born and no evidence of adverse effects on development. Moreover, it became evident that pregnancy rates after PGD were comparable to those obtained after standard IVF.
Despite the reassuring clinical results, there remains a question regarding the possibility of reduced reproductive potential after a cleavage-stage biopsy. Initial studies showed no impact with one-cell removal, but that removal of two cells from a seven-cell–stage embryo (or larger) reduces its capacity to implant more than if only one cell were removed. Owing to the limited genetic material provided by a single cell, two cells were often removed from the embryo. Follow-up studies on whether cleavage-stage biopsies were harmful to embryo implantation potential were inconsistent. A randomized controlled trial showed that removal of two blastomeres significantly decreases the likelihood of blastocyst formation compared with removal of one blastomere, although this did not translate into a significant difference in implantation rate of a fresh day-5 transfer (23.5% vs. 17.3% for one-cell and two-cell biopsy, respectively, P = .216). Taking live births per started cycle as an end point of this study (including 592 biopsy cycles), no significant difference was obtained between one-cell (20.2%) and two-cell biopsy (17.2%, P = .358). However, most recently, in a more eloquent paired randomized controlled trial, it was found that a cleavage-stage biopsy may be associated with a reduction in sustained implantation and, consequently, live birth.
One possible way to estimate the effect of removing blastomeres from cleavage-stage embryos can be derived from experience with cryopreservation at the cleavage stage and associated cell loss. It has been shown that loss of viability after freezing is proportional to the number of blastomeres that lyse during the freezing and thawing process. For example, if the expected implantation is 15%, then the loss of one blastomere from an eight-cell embryo would result in an implantation equal to 7/8 × 15 = 13.1%. Likewise, the removal of two blastomeres from the same embryo would result in an implantation equal to 6/8 × 15 = 11.3%.
In summary, the actual impact of a cleavage-stage biopsy on reproductive potential appears to be related to the proportion of material that is removed from the embryo. Therefore the current recommendation for cleavage-stage biopsies is removal of only one blastomere provided that diagnostic safety measures are in place to ensure a correct diagnosis on one cell. If these are not available, two cells will be needed for diagnosis.
Cleavage-stage biopsies for PGD are highly accurate. As in the case of polar-body biopsies, the detection of genetic disorders requires amplification of the DNA in combination with multiplex PCR to increase accuracy and detect possible allele dropout and contamination. However, in contrast to polar-body biopsy, both maternal and paternal meiotic errors can be detected and there is no need for sequential biopsies to increase accuracy. Some clinics may consider proposing PGD at this stage rather than the blastocyst stage in poor-prognosis patients to increase the possibility of detecting a normal embryo (more embryos to biopsy) given the accuracy at this stage and the high arrest rate of embryos with extended culture.
PGS results with cleavage-stage biopsies are difficult to interpret. Embryos at the cleavage stage have a high degree of chromosome instability. As a result, mitotic errors are high. In fact, the incidence of mitotic errors in cleavage-stage embryos is higher than the meiotic aneuploidy rate; as a consequence, the genome of a single blastomere may not be representative of the genome of the other cells in the embryo. Because of the frequency of mitotic errors, over half the embryos at this stage are mosaic , which is defined as the presence of two or more chromosomally distinct cell lines within an embryo. Therefore day-3 embryos can consist of a mixture of normal cells and cells with aneuploidy. Mitotic errors are most commonly due to anaphase lag and nondisjunction, concepts discussed in detail in a review by Mantikou and collegues. Multiple types of mosaicism are characterized, some with undetermined significance. Contributing to the challenges of assessing PGS outcomes, it is also thought that many of these cleavage-stage embryos diagnosed as aneuploidy due to mitotic errors will self-correct by the blastocyst stage. The concept of self-correction, albeit controversial, is based on the relatively higher proportion of aneuploid cells present within the embryo at the cleavage stage versus the blastocyst stage and evidence for trisomic rescue.
Prior randomized trials with cleavage-stage biopsies for PGS showed no benefit. The main reason for the surprising findings was due to the mosaicism and consequently high number of false-positive and false-negative results. In addition, FISH was used to test chromosome copy number, which was complicated by a low predictive value and limitations as to the number of chromosomes assessed (see further on). With newer molecular testing and the ability to do CCS, it is possible that results would lead to increased diagnostic challenges given the sensitivity of the testing. More research is required to understand the clinical consequences of chromosome instability and mosaicism.
Cleavage-Stage Biopsy Procedure
Cleavage-stage biopsy is performed on the morning of day 3 after oocyte retrieval ( Fig. 32.13 ). Unfortunately not all human embryos reach the seven- or eight-cell stage by that time. Six-cell–stage embryos may, therefore, be included for one- or two-cell removal, although they are suspected of being of inferior quality because of delayed cleavage.
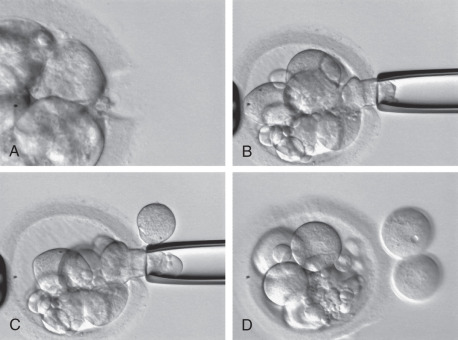
Related posts:
 Gonadotropin Hormones and Their Receptors
Gonadotropin Hormones and Their Receptors
 Steroid Hormones and Other Lipid Molecules Involved in Human Reproduction
Steroid Hormones and Other Lipid Molecules Involved in Human Reproduction
 Structure, Function, and Evaluation of the Female Reproductive Tract
Structure, Function, and Evaluation of the Female Reproductive Tract
 Endocrine Disturbances Affecting Reproduction
Endocrine Disturbances Affecting Reproduction
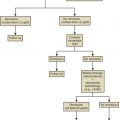 Physiologic and Pathophysiologic Alterations of the Neuroendocrine Components of the Reproductive Axis
Physiologic and Pathophysiologic Alterations of the Neuroendocrine Components of the Reproductive Axis
 Pelvic Imaging in Reproductive Endocrinology
Pelvic Imaging in Reproductive Endocrinology
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


