Familial Multiple Coagulation Factor Deficiencies
Bin Zhang
David Ginsburg
The familial multiple coagulation factor deficiencies (FMFDs) are a group of rare inherited disorders characterized by a simultaneous decrease in the levels of two or more coagulation factors. Earlier reports of deficiencies in various combinations of coagulation factors prompted Soff and Levin1 in 1981 to classify these disorders into six types (FMFD I to VI). Combined deficiency of FV and FVIII (F5F8D, or FMFD I) and combined deficiency of vitamin K-dependent clotting factors (VKCFD, or FMFD III) are the best known and comprise the vast majority of reported cases of FMFD. This chapter focuses on these two disorders. Little is known about other types of multiple coagulation factor deficiencies, as few new cases have been reported since 1981. As a consequence, this old classification is of limited relevance today and not generally used in practice.2
COMBINED DEFICIENCY OF FACTOR V AND FACTOR VIII
History
Combined deficiency of factors V and VIII (F5F8D) was first described in a pair of Swiss siblings in 1954.3 Since then, more than 80 families with F5F8D have been identified around the world. As is the case for rare autosomal recessive disorders, F5F8D is often associated with consanguineous marriages. However, it is likely that F5F8D is underdiagnosed, in part due to its often mild bleeding manifestations. Some F5F8D patients may be misdiagnosed as having mild hemophilia A or parahemophilia (FV deficiency). F5F8D appears to be particularly prevalent among Middle Eastern Jews and non-Jewish Iranians, estimated at approximately 1:100,000 in these populations.4,5,6 This high frequency is probably due, at least in part, to the prevalence of consanguineous marriages in these populations. The molecular basis of F5F8D was a mystery until 1998.7 Initial speculation centered on a defect in a precursor protein for both factors V and VIII,3 but no such precursor could be identified. A low level of protein C inhibitor activity in patient plasma was reported in 1980,8 but was later found to be caused by an artifactual loss of protein C inhibitor in frozen samples.9,10,11,12 Subsequent genetic studies identified the cause of this disorder as deficiencies in a unique secretory pathway shared by FV and FVIII.13,14,15
Genetics and Biology
Inheritance of F5F8D is autosomal recessive and distinct from the coinheritance of both FV deficiency and FVIII deficiency. Heterozygotes are asymptomatic and their plasma levels of FV and FVIII are indistinguishable from normal. Using homozygosity mapping and positional cloning approaches, the first gene for F5F8D was identified in 199813 as LMAN1 (lectin, mannose-specific 1). The encoded protein (LMAN1, also referred to as ERGIC-53), though of previously unknown function, was known to localize to the endoplasmic reticulum (ER)-Golgi intermediate compartment (ERGIC).16,17 Mutations in LMAN1 account for approximately 70% of F5F8D patients. No LMAN1 mutations could be identified in approximately 30% of affected families.18,19 In 2003, the second cause of F5F8D was identified in the latter subset of families as mutations in MCFD2 (multiple coagulation factor deficiency gene 2).14 To date, at least 33 different LMAN1 mutations13,18,19,20,21 and 17 different MCFD2 mutations14,21,22,23 have been identified (FIGURE 55.1). The diverse nature of the mutations indicates multiple independent genetic origins. Haplotype analyses performed on two MCFD2 mutations (c.149+5G>A and c.407T>C) that are identified in multiple distinct populations demonstrated multiple independent genetic origins for each mutation.24 However, founder mutations may account for all or the majority of F5F8D in some isolated populations. For example, two mutations in LMAN1 account for nearly all Jewish F5F8D patients. One of these mutations is prevalent in Jews originating from the island of Djerba in Tunisia, but not found among North African Jews.25 The second LMAN1 mutation is unique to Middle Eastern Jews.13 Another notable mutation is the missense mutation abolishing the initiation codon (c.2T>C) of LMAN1, which has only been reported in families of Italian origin.21,26
Mean FV and FVIII levels are slightly but significantly lower in MCFD2 patients compared to LMAN1 patients. However, the levels in these two groups overlap extensively, and individual patients with MCFD2 or LMAN1 mutations are clinically indistinguishable.21 Plasma FV and FVIII levels for a given patient are also significantly correlated, suggesting that deficiencies in either LMAN1 (ERGIC-53) or MCFD2 exert a similar impact on the secretion of both FV and FVIII.21 Circulating FV exists in both plasma and the α-granules of platelets as a 330-kDa single-chain polypeptide. In humans, the platelet FV pool is thought to originate from endocytosis of plasma FV,27,28 whereas in mice, platelet FV is derived exclusively from biosynthesis within the megakaryocytes.29,30 Comparison of platelet FV levels in F5F8D patients and normal controls indicates that platelet FV and plasma FV are reduced to the same extent for both the LMAN1 group and the MCFD2 group.21 Therefore, mutations in LMAN1 or MCFD2 similarly affect the steadystate levels of FV in both pools.
LMAN1 is a 53-kD homohexameric transmembrane protein that belongs to a conserved class of L-type animal lectins with homology to leguminous mannose-binding lectins.31,32 The
LMAN1 C-terminus is exposed to the cytoplasm and contains the ER exit and retrieval motifs. The ER luminal side of the protein contains a carbohydrate recognition domain (CRD) and a coiled-coil domain. MCFD2 is a 16-kDa soluble protein with two Ca2+-binding motifs, known as EF-hand domains, at the C-terminus with a structure similar to that of calmodulin.33 MCFD2 interacts with LMAN1 to form a stable Ca2+-dependent complex with a 1:1 stoichiometry, and the two proteins colocalize in the ERGIC.34 Crystal structure and cell biology studies indicate that the N-terminus of the LMAN1 CRD interacts with the EF-hand domains of MCFD2.35,36 Only three missense mutations have been identified in LMAN1. The first missense mutation destroys the initiation codon of the gene. The other two mutations either destabilize the protein24 or abolish MCFD2 binding.20 Seven MCFD2 missense mutations have been identified within the EF-hand domains, which disrupt the LMAN1-MCFD2 interaction, resulting in F5F8D.14,21,37
LMAN1 C-terminus is exposed to the cytoplasm and contains the ER exit and retrieval motifs. The ER luminal side of the protein contains a carbohydrate recognition domain (CRD) and a coiled-coil domain. MCFD2 is a 16-kDa soluble protein with two Ca2+-binding motifs, known as EF-hand domains, at the C-terminus with a structure similar to that of calmodulin.33 MCFD2 interacts with LMAN1 to form a stable Ca2+-dependent complex with a 1:1 stoichiometry, and the two proteins colocalize in the ERGIC.34 Crystal structure and cell biology studies indicate that the N-terminus of the LMAN1 CRD interacts with the EF-hand domains of MCFD2.35,36 Only three missense mutations have been identified in LMAN1. The first missense mutation destroys the initiation codon of the gene. The other two mutations either destabilize the protein24 or abolish MCFD2 binding.20 Seven MCFD2 missense mutations have been identified within the EF-hand domains, which disrupt the LMAN1-MCFD2 interaction, resulting in F5F8D.14,21,37
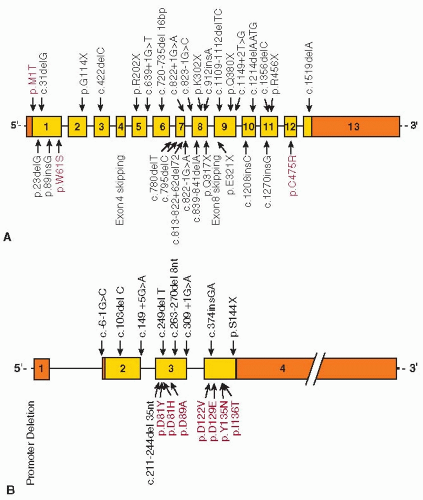 FIGURE 55.1 Structures of LMAN1 and MCFD2 genes and mutations identified in combined deficiency of factor V and factor VIII. A: Most mutations in LMAN1 are predicted to result in complete loss of protein function. Three missense mutations, shown in red, also lead to loss of function. B: In addition to insertion, deletion, and splice site mutations in MCFD2, multiple missense mutations also occur in both EF-hand domains and were shown to abolish binding to LMAN1(14;21;37). Exons are numbered and drawn to scale. Shaded rectangles represent the coding regions of the genes, with the open portions of exons 1 and 13 in LMAN1 and exons 1, 2, and 4 in MCFD2 representing the 5′ and 3′ untranslated regions. |
One of the central problems in cell biology is how newly synthesized proteins are sorted for transport to their final destinations. An increasingly accepted model envisions that “cargo receptors,” localized to the ER membrane, selectively package secreted proteins into “vesicles” for transport to the Golgi body.38,39 The finding of LMAN1 gene mutations as a cause of F5F8D provided the first direct evidence for a mammalian ER cargo receptor, suggesting that LMAN1 mediates export of FV and FVIII from the ER to the Golgi (FIGURE 55.2). To date, the LMAN1-MCFD2 complex remains the only cargo receptor identified in higher eukaryotes,38,39 though two other potential cargo receptors have been described in yeast.40,41 LMAN1 and MCFD2 should act in concert to facilitate transport of FV and FVIII, since mutations in either gene lead to impaired secretion of FV and FVIII. However, the precise role of MCFD2 in cargo receptor function is still not clear. Both LMAN1 and MCFD2 have been shown to bind FV and FVIII directly,14,34,37,42 with the sugar-binding site of LMAN1 and the EF-hand domains of MCFD2 important for these interactions.37,42 The heavily glycosylated B domains of FV and FVIII may be important in mediating secretion through the LMAN1-MCFD2 pathway.34,43,44 An interesting question is whether the ER to Golgi transport of other proteins is also altered in F5F8D patients. This seems likely, given the ubiquitous expression patterns of both LMAN1 and MCFD2,14 as well as the presence of LMAN1 and MCFD2 orthologs in lower eukaryotes without a blood clotting system. Indeed, a number of other proteins have been identified to either interact with LMAN1 or exhibit delayed secretion in cultured cells with a
dysfunctional LMAN1 (FIGURE 55.2).45,46,47,48 However, transport deficiency in other proteins is likely to be at a level insufficient to produce a clinical phenotype, as no other abnormalities, aside from the deficiencies of FV and FVIII, have been identified in F5F8D patients.
dysfunctional LMAN1 (FIGURE 55.2).45,46,47,48 However, transport deficiency in other proteins is likely to be at a level insufficient to produce a clinical phenotype, as no other abnormalities, aside from the deficiencies of FV and FVIII, have been identified in F5F8D patients.
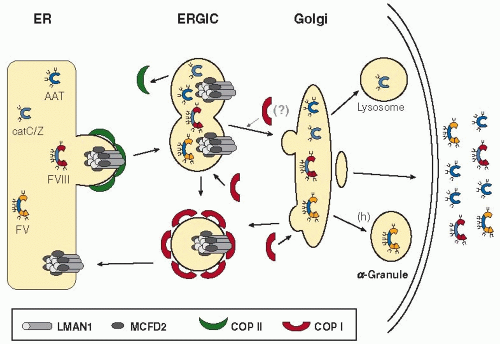 FIGURE 55.2 Model of receptor-mediated ER to Golgi transport of FV and FVIII and other potential cargo. Correctly folded FV/FVIII molecules are recruited to the COPII-coated vesicle budding from the ER by binding to the LMAN1-MCFD2 complex. Upon budding, these vesicles uncoat and fuse with each other to form the ERGIC. Release of FV/FVIII from LMAN1-MCFD2 occurs in the ERGIC. The LMAN1-MCFD2 complex is recycled back to the ER in COPI-coated retrograde vesicles as FV and FVIII are transported to the Golgi. Cargo proteins undergo further sorting and modifications in the Golgi. CatC and catZ are targeted to the lysosome. FV, FVIII, and α1-antitrypsin (AAT) are secreted from cells, with the exception of megakaryocytes, in which FV is packaged into α-granules for storage. (Modified by permission from Baines AC, Zhang B. Receptor-mediated protein transport in the early secretory pathway. Trends Biochem Sci 2007;32:381-388, copyright (2007) Elsevier Ltd.) |
Clinical Manifestations
Seligsohn et al.4 studied 14 patients in 7 families of Middle Eastern Jews. Peyvandi et al.5 and Mansouritorgabeh et al.6 studied a total of 46 patients from 23 families of non-Jewish Iranians. In addition to these relatively large studies, a number of case reports have also been published.23,26,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76 Patients with this disorder exhibit plasma FV and FVIII activity level at approximately 10% of normal, with a range of 5% to 30%, though factor levels as low as 1% have been reported.5,49 FV and FVIII clotting activity and antigenic level are both concordantly decreased.1,67,77 Bleeding symptoms appear to be comparable to those observed in patients with single deficiencies of FV or FVIII.4,5 No abnormalities outside of these hemostatic changes have been identified in F5F8D. Spontaneous bleeding symptoms are common, including easy bruising, epistaxis, and menorrhagia. Hemarthroses are less frequent, though reported in up to one-third of patients in some series.5,6,49 In contrast, gastrointestinal bleeding, hematuria, and intracranial bleeding are only rarely observed. Excessive bleeding is particularly common during or after trauma, tooth extraction, surgery, or labor. Postcircumcision bleeding was reported to be common among Iranian patients,5 but rare among Jewish patients,4 possibly due to the age at which circumcision is performed (at birth among Jews and usually at 5 to 7 years of age among Muslims). In contrast to hemophilia A, the levels of FV and FVIII correlate poorly with bleeding severity.
Related posts:
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Integrín αIIbβ3 and Platelet Aggregation
Integrín αIIbβ3 and Platelet Aggregation
 Acquired Nonimmune Thrombocytopenia
Acquired Nonimmune Thrombocytopenia
 Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


