Visit the Endocrine Hypertension: From Basic Science to Clinical Practice , First Edition companion web site at: https://www.elsevier.com/books-and-journals/book-companion/9780323961202 .



Introduction
Primary aldosteronism (PA), the most common cause of endocrine hypertension, is characterized by autonomous overproduction of aldosterone from the adrenal gland(s). Of the several subtypes of PA, bilateral adrenal hyperplasia or idiopathic hyperaldosteronism accounts for ∼60% of cases and aldosterone-producing adenomas (APAs) ∼30% of cases, with unilateral hyperplasia (less common), malignancy, and familial hyperaldosteronism (FH; both rare) forming the remainder [ , ]. The genetic landscape of PA has been well characterized in recent years, and somatic mutations have been identified in more than 90% of APAs [ , ]. Germline mutations are also described in sporadic PA with bilateral adrenal hyperplasia making our understanding of the disease more complex.
FH is a group of rare disorders causing monogenic hypertension, and all these are inherited in an autosomal dominant manner [ ]. Although genotypically distinct often with a positive family history, FH is characterized by a mineralocorticoid excess state (as in sporadic PA) with early-onset refractory hypertension, hypokalemia, metabolic alkalosis, raised aldosterone–renin ratio (ARR), and probable hypertension-related cardiovascular complications. An accurate diagnosis requires genetic testing, as clinically and biochemically these disorders are often indistinguishable from sporadic PA [ ]. This chapter is to equip the readers with the current evidence-based scientific knowledge on these rare genetic disorders causing endocrine hypertension.
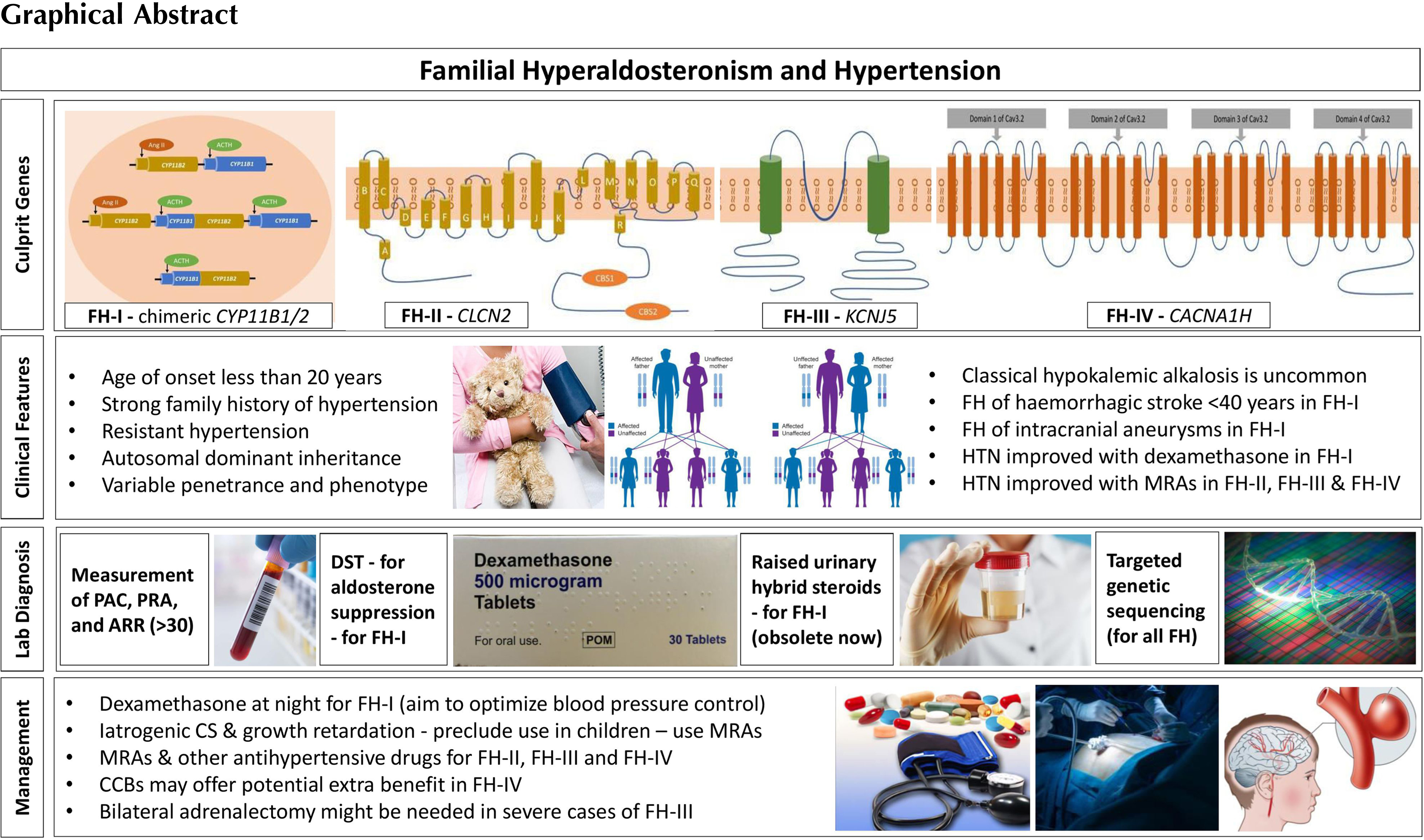
Pathophysiology of familial hyperaldosteronism
Depending on the genetic characteristics, molecular features, and pathophysiological aspects, FH is classified into four major subcategories (FH-I, II, III, and IV). In addition, a complex syndrome of familial PA with neurological abnormalities such as cerebral palsy and seizures known as PASNA (primary aldosteronism, seizures, and neurological abnormalities) syndrome has also been described [ , ]. Genetic testing for FH is recommended in an individual with biochemically proven mineralocorticoid excess when there is a family history of PA with early onset of hypertension [ ]. A detailed review of the pathophysiological aspects, clinical profiles, and management options of individual disorders is discussed under the subtypes of FH below.
Subtypes of familial hyperaldosteronism
Familial hyperaldosteronism type 1
Pathophysiology: FH-I, also known as glucocorticoid-remediable aldosteronism (GRA), is the most common form of monogenic hypertension [ ]. FH-I was first identified in 1966 by Sutherland et al. in a father and son presenting with hypertension, elevated aldosterone, suppressed renin, and metabolic alkalosis that improved with dexamethasone treatment [ ]. This genetic disorder occurs as a result of an unequal cross-over between the neighboring and highly homologous CYP11B1 and CYP11B2 genes on chromosome 8q24.3, generating a chimeric variant gene [ ]. The CYP11B1 gene encodes 11β-hydroxylase. It is expressed under the control of ACTH in the adrenal zona fasciculata. The CYP11B2 encodes aldosterone synthase, which differs from 11β-hydroxylase in that it possesses an 18-hydroxylase activity in addition to the 11β-hydroxylase activity. Its expression is triggered by both angiotensin II and hyperkalemia and is confined normally to the zona glomerulosa. The unequal cross-over event results in an enzyme with aldosterone synthase activity but expressed under the control of ACTH in the zona fasciculata. The ectopic expression of an enzyme with 18-hydroxylase activity in the zona fasciculata results in the synthesis of aldosterone and other hybrid steroids such as 18-oxocortisol and 18-hydroxycortisol. The conversion of cortisol to 18-oxocortisol and 18-hydroxycortisol leads to decreased cortisol secretion, triggering further ACTH production, and as such, mineralocorticoid levels are often grossly elevated in FH-I cases [ , , ]. FH-I shows an autosomal dominant inheritance pattern with variable penetrance that results in milder forms of the disease in a proportion of patients [ , , ]. Fig. 8.1 shows the schematic representation of the pathophysiological aspects of FH-I.
Clinical features: Classical clinical picture of patients with FH-I is early-onset hypertension with a strong family history and refractory hypertension not amenable to the standard antihypertensive medications [ ]. In a large retrospective study reviewing 376 patients from an international registry of FH-I, 18% had cerebrovascular accidents (CVAs) [ ]. This cohort included 27 genetically proven FH-I pedigrees in whom the incidence of CVA was 48%. Of these, 70% were hemorrhagic strokes with an overall case fatality rate of 61%. Intracranial aneurysms were the cause of brain hemorrhage, the prevalence of which appeared to be comparable to that in adult polycystic kidney disease [ ]. Variable degree of penetrance of the genetic defect may result in phenotypic variations with mild hypertension or even normotension in some individuals with FH-I [ ]. There is an increased risk of preeclampsia in pregnant women with FH-I [ ].
Diagnostic evaluation: Patients with onset of PA before the age of 20 years with a family history of PA or stroke at an age below 40 years should be tested for FH-I as per the current guidelines of the Endocrine Society [ , , ]. The initial diagnosis is based on the biochemical confirmation of PA. Although hypertension with hypokalemic alkalosis is the classical description of PA, plasma potassium levels are often normal with hypokalemia an infrequent feature (≈20%) except when diuretics are used. Initial screening is with estimation of plasma aldosterone concentration coupled with plasma renin activity as per the standard testing protocol to obtain ARR [ , ]. An ARR cut off >30 in a patient with clinical suspicion of the disease has a high sensitivity for PA. Biochemical confirmation of PA is as per the standard recommendations with special precautions taken to avoid fluid overload or hypertension-related complications and is described in Chapter 7, Chapter 10, Chapter 21 .
Once the diagnosis of PA is established, genetic testing for the chimeric CYP11B1/2 gene can be performed for confirmation of FH-I. A low-dose dexamethasone suppression test (DST) for suppression of plasma aldosterone levels to <4 ng/dL (<110 pmol/L) and/or >80% suppression from baseline after oral dexamethasone 0.75–2 mg daily for 2 days has very high sensitivity and specificity for biochemical evidence of FH-I that also confirms the ACTH control of aldosterone overproduction in these patients and has a high correlation with genetic testing [ ]. Patients with FH-I also excrete large amounts of urinary hybrid steroids including 18-hydroxycortisol and 18-oxocortisol that were used for the diagnostic screening in the past along with their suppression following DST [ ]. However, these biochemical screening tests are largely obsolete now with the advent of targeted genetic sequencing methods for the culprit gene in the recent years, coupled with the finding that the hybrid steroids are also observed in other familial forms of aldosteronism [ ].
Management: Usual antihypertensive agents are less likely to be effective in patients with FH-I. Low doses of oral steroids to optimally suppress the ACTH to keep the levels of aldosterone and hybrid steroids normal usually help to control hypertension in these patients. Dexamethasone 0.125–0.250 mg administered at night to suppress the normal early morning surge of ACTH will usually control the disease in patients with FH-I [ , ]. Dexamethasone is preferred because of its least mineralocorticoid activity among the oral corticosteroids and a less preferred alternative is prednisolone at the dose of 2.5–5 mg daily. Iatrogenic Cushing’s syndrome and growth retardation may preclude the use of steroids among children in whom mineralocorticoid receptor (MR) antagonists such as spironolactone or eplerenone can be effective alternatives to control hypertension [ , , , ]. The target of treating patients with FH-I should be the optimal control of blood pressure rather than the normalization of elevated mineralocorticoids to avoid undesirable side effects of long-term steroids in these patients [ ].
Familial hyperaldosteronism type 2
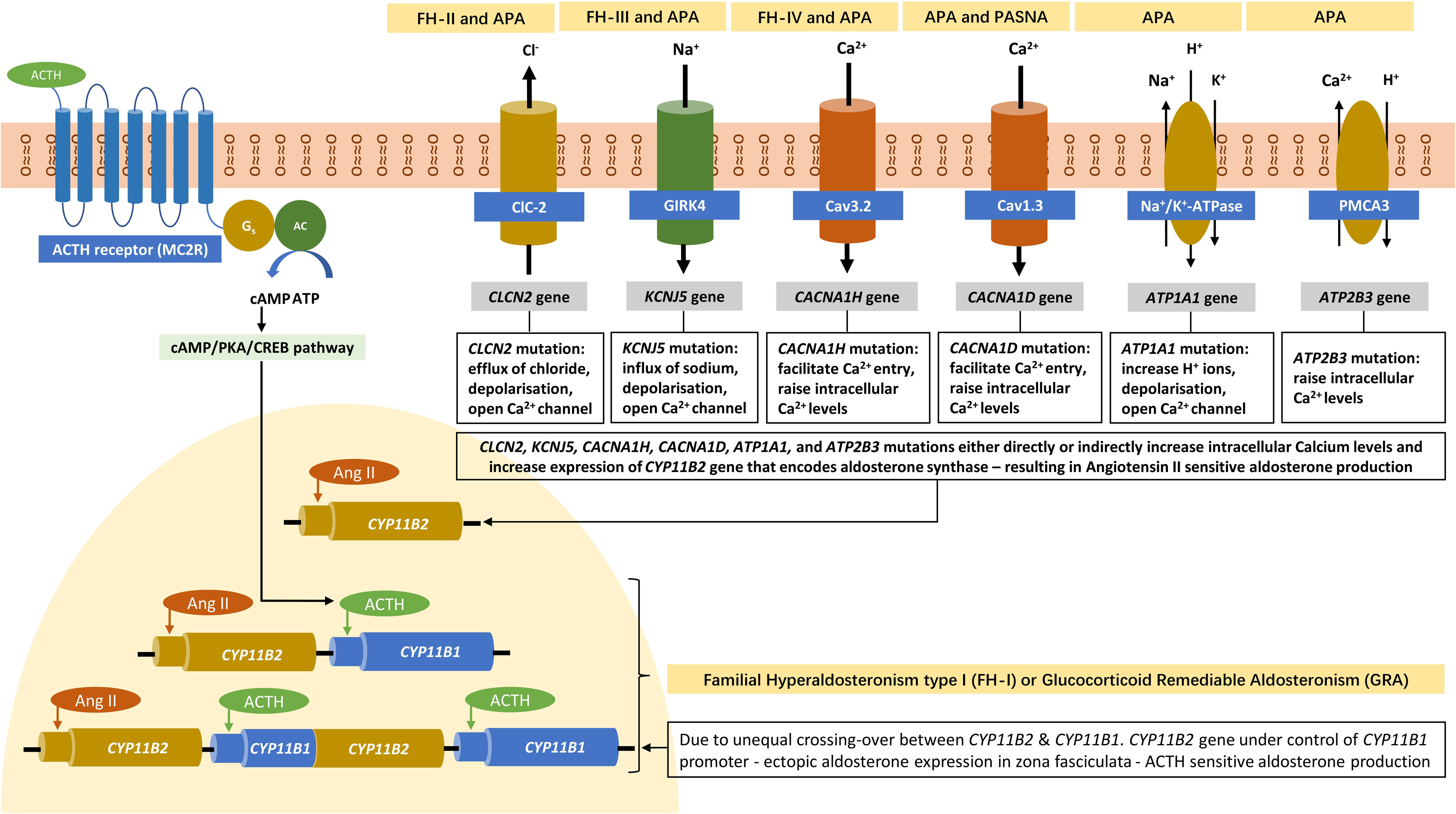
Pathophysiology: FH-II was first described by Stowasser et al. among 13 patients from 5 families in whom FH was not suppressible with dexamethasone and these cases also had a high tendency for APA formation [ ]. The culprit gene associated with FH-II is a gain-of-function mutation in CLCN2 gene with variable penetrance that encodes a chloride channel ClC2 as demonstrated by Scholl et al. in 2018 [ ]. The mutation was identified from the DNA analysis by exome sequencing of three patients from one of the families with FH-II originally described by Stowasser et al. in 1992. The mutant variant p.Arg172Gln was also shown in five other family members of the original cohort [ ]. CLCN2 gene is located on chromosome 3q27 and the voltage-gated chloride channel (ClC2) is widely expressed in mammalian tissues such as the brain, heart, gastrointestinal tract, kidney, adrenal glands, and the liver [ , ]. When the ClC2 opens (with a higher tendency among CLCN2 mutant genes), it depolarizes adrenal zona glomerulosa cells inducing aldosterone synthase (the rate-limiting enzyme for aldosterone synthesis) expression, resulting in overproduction of aldosterone [ ]. The mineralocorticoid excess state, as a consequence, results in hypertension that is indistinguishable from other sporadic forms of PA biochemically. CLCN2 somatic mutations are also described in APAs and bilateral adrenal hyperplasia, though FH-II almost always presents in childhood or adolescence with germline mutations in these patients. Fig. 8.1 demonstrates the pathophysiology of FH-II.
Clinical features: FH-II is clinically indistinguishable from the sporadic forms of PA except for the early age of onset as mentioned above. As in FH-I, the disease severity can vary depending on the penetrance of the genetic mutation [ , ]. Early-onset hypertension with typical age at diagnosis before 20 years is characteristic. Classical clinical presentation “hypokalemic metabolic alkalosis” is uncommon. Although this form of FH has been considered the most common type in the past, recent identification of the culprit gene has changed this notion as many of the case with a diagnosis FH-II would have been cases with sporadic PA [ ].
Diagnostic evaluation: Once PA diagnosis is established in a child or adolescent with hypertension as described above, genetic testing is recommended in patients with a family history of PA. DST is not useful for the biochemical identification of the disease. Targeted gene sequencing for CLCN2 would help to establish the diagnosis [ , , ]. Screening among first-degree relatives with hypertension should be considered as per the Endocrine Society guidelines as we are still unaware of the exact prevalence of the disease and the degree of penetrance of the genetic defect [ ].
Management: Hypertension in patients with FH-II may respond to MR antagonists and other antihypertensive agents [ ]. However, there is inadequate global expertise on the long-term outcome of such a strategy owing to inadequate experience in managing such patients.
Familial hyperaldosteronism type 3
Pathophysiology: In 2008, Geller and colleagues described a new variant form of FH in a father and 2 daughters with severe refractory hypertension diagnosed at the age of 7 years [ ]. These patients had high levels of hybrid steroids 18-oxocortisol and 18-hydroxycortisol, along with hyporeninemic hyperaldosteronism without suppression of aldosterone, cortisol, and hypertension on DST as expected in patients with FH-I. This autosomal dominant disease without any abnormalities in the aldosterone synthase gene and the abnormal biochemical profile implied a global defect the adrenal steroidogenesis pathway. Patients had to undergo bilateral adrenalectomy for the cure of severe and resistant hypertension [ ]. The adrenal gland specimens of these cases revealed massive hyperplasia with a disorganized zonation pattern, and an enlargement of the zona fasciculata and transitional zone, with an atrophic zona glomerulosa layer. This disorganized zonation characteristic with the presence of cells that coexpress enzymes which are usually expressed in the distinct individual zones of the adrenal cortex, such as CYP11B1 and CYP11B2, and also CYP17A1, might explain abnormally exaggerated steroidogenesis including that of hybrid mineralocorticoids like 18-oxocortisol and 18-hydroxycortisol [ , , ].
The culprit gene, later sequenced by Choi et al. in 2011 on chromosome 11q24, is a heterogenous gain-of-function mutation in KCNJ5 that encodes a G-protein-coupled potassium channel 4 (Kir3.4) [ , ]. Kir3.4 with other Kir proteins controls the polarization of the zona glomerulosa cell membranes. The mutations described by Choi in KCNJ5 result in a loss of channel specificity, allowing sodium to pass through this potassium channel from which it is normally excluded. Sodium current through the channel results in depolarization of the cell, triggering a voltage-sensitive calcium current which leads to overproduction of aldosterone [ ]. Several mutations in the KCNJ5 gene were described subsequently and only those familial cases with germline mutations are categorized as FH-III [ , , , ]. Somatic mutations of KCNJ5 gene are also commonly seen in patients with APAs and bilateral adrenal hyperplasia associated with PA [ , ]. The pathophysiological aspects of FH-III are shown in Fig. 8.1 .
Clinical presentation of FH-III: The disease is rare accounting for <1% of cases with PA [ ]. The phenotype of FH-III patients varies widely with the mutation. The family described by Geller et al. harboring the T158A allele had severe disease requiring bilateral adrenalectomy [ ]. A similar phenotype was reported for patients harboring a G151R allele [ ]. In contrast, Scholl et al. reported that patients with a G151E mutation (a mutation not seen in APAs) had mild, easily controlled disease. Interestingly, the G151E mutation had a greater effect on loss of channel specificity; the relatively free flow of sodium into the cell leads to cell lethality, with the inference that this lethality limits adrenal cell mass and thus aldosterone production. It is a striking example of how small amino acid changes can result in markedly different phenotypes.
Diagnostic evaluation: Clinical picture and biochemical features of PA described earlier with a very early onset of hypertension in childhood may alert clinicians for a diagnostic evaluation with genetic testing. DST does not suppress the mineralocorticoid excess and improve hypertension. There may be a paradoxical rise in blood pressure and mineralocorticoid levels to DST as observed by Geller et al. in their cases [ ]. The Endocrine Society suggests genetic testing for FH-III in patients with very early onset of hypertension with positive family history in PA [ ].
Management: Mild forms of the disease may be managed with MR antagonists such as spironolactone or eplerenone [ , ]. However, severe cases may need bilateral adrenalectomy for control of the disease and hypertension [ , , , ]. Bilateral adrenalectomy is curative in severe cases, but triggers the need for lifelong steroid replacement therapy; early adrenalectomy presumably reduces the risk of cardiovascular sequelae of the aldosteronism, such as left ventricular hypertrophy, which was present in the young women described by Geller et al. [ ].
Familial hyperaldosteronism type 4
Pathophysiology: FH-IV was first reported by Scholl et al. among five unrelated patients with early onset PA (≤10 years of age) in a cohort of 40 patients without any known PA genes [ ]. By genome sequencing they found a gain-of-function mutation (p.Met1549Val) in the CACNA1H gene that encodes a T-type calcium channel subunit (Ca v 3.2) detected on chromosome 16p13 to cause the genetic anomaly. This abnormal Ca 2+ channel causes increased calcium influx in the adrenal cortical cells, perhaps secondary to altered sodium conductivity of the mutant channel, but ultimately resulting in aldosterone overproduction [ , ]. CACNA1H is the second most expressed gene in the zona glomerulosa cell layer of adrenal gland for the synthesis of calcium channels [ , ]. Several other mutations have been subsequently described in this gene accounting for somatic and germline defects associated with PA [ , ]. Only those with germline mutations and heritability are classified as FH-IV, while the others with somatic gene defects may present with APAs. A schematic diagram explaining the pathophysiology of FH-IV is demonstrated in Fig. 8.1 .
Clinical profile: Although FH-IV is an autosomal dominant genetic disorder, because of variable penetrance the clinical spectrum may range from mild disease expression (mild hypertension or even normotension) to severe and treatment resistant hypertension from early life [ ]. In the patients described by Scholl et al., all children had clinical presentations suggestive of hyperaldosteronism, but adult carriers did not, suggesting decreased disease penetrance by age. The reasons for this are not clear.
Diagnostic evaluation: Patients presenting with PA in childhood and a family history of the disease should be tested for FH-IV as per the current Endocrine Society recommendations [ ].
Management: MR antagonists such as spironolactone and eplerenone are useful for the management of hypertension in patients with FH-IV [ , ]. Calcium channel blockers have been found to inhibit Ca 2+ currents through CACNA1H and thus may be of use as well, but there is not much experience with these agents because of the rarity and discovery of the disorder only in the recent years [ ]. The long-term prognosis of the condition remains elusive for the same reasons.
PASNA syndrome
A rare and complex disorder comprising PA, seizures, and neurological abnormalities (PASNA) was described with de novo heterozygous gain-of-function mutations in the CACNA1D gene encoding an abnormal calcium channel (Ca v 1.3—a high voltage gated Ca 2+ channel) among two individuals with cerebral palsy in 2013 [ ]. However, expression of this channel in other organs such as the brain, heart, and pancreas without PA was identified later, and as such, this genetic defect is not considered as a classical form of FH [ , ]. Other CACNA1D mutations without PA also have been described [ ].
Emerging research questions/future research
Genetic studies of PA are still evolving with newer genes identified in several somatic and germline defects identified in the recent years. More familial disorders causing PA are likely to emerge in the coming years. Germline mutations in the ARMC5 , a tumor suppressor gene, have been found to be associated with PA recently [ ]. It is not clear whether these would fall into the category of Familial PA in future with more cases and heritability of this entity described with evolving research. Rare germline variants of the phosphodiesterase 2A ( PDE2A ) and 3B ( PDE3B ) genes were identified from the unilateral adrenalectomy specimens in three patients with early onset hypertension and PA from bilateral adrenal hyperplasia [ ]. We are unsure about the familial nature of these cases also, and future research should shed more light on the genetic landscape of PA for our better understanding of the disease.
Summary and conclusions
FH is associated with early-onset monogenic hypertension with a strong family history and accounts for only a small proportion of patients with PA. The disease severity may vary depending on the degree of penetrance of the FH causing gene disorder. GRA is the most common form of monogenic hypertension. Diagnostic work up of FH involves biochemical confirmation of PA followed by appropriate molecular genetic analysis to identify the culprit gene causing the disease. Newer genes associated with both familial and sporadic PA are still being discovered improving our understanding of these uncommon genetic disorders.
Learning points
- •
FH is a group of genetic disorders associated with childhood-onset monogenic hypertension and with a strong family history.
- •
FH-I or GRA is caused by an unequal cross-over between the CYP11B1 and CYP11B2 genes on chromosome 8q24.3, generating a chimeric gene that results in ACTH-dependent aldosterone production in other layers of adrenal cortex apart from zona glomerulosa. The disease therefore can be controlled by ACTH suppression with glucocorticoids.
- •
Phenotypic variations can occur in the disease severity in patients with FH depending on the gene penetrance in the affected individuals. On occasions, bilateral adrenalectomy may be necessary to control hypertension in severe forms of FH-III.
References
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


