Introduction
The diagnosis and management of familial endocrine syndromes epitomises the complex and changing interface between surgery, medicine and molecular genetics. The last decade has seen an explosion in our understanding of the molecular basis of these rare syndromes, and the rapid translation of research-based findings into clinical practice. As a result, genetic testing is already resulting in highly effective, targeted intervention. The next decade is likely to see continued progress, with expansion and refinement of molecular diagnostics and further integration of these developments into clinical practice. We must be mindful, however, of the limitations of molecular medicine, and the ethical context in which molecular medicine should be practised.
This chapter will cover the genetics, presentation and management of a range of conditions relevant to endocrine surgical practice. This is a complex clinical area, and one that encompasses several professional boundaries and the interface between paediatric and adult medicine. A coordinated and integrated approach is essential.
A brief overview of clinical endocrine genetics
The growth, replication and differentiation of cells are regulated by many different genes. When these genes become damaged – or ‘mutated’ – cell proliferation may become disordered and give rise to a tumour, whether benign or malignant. The majority of tumours result from acquired genetic damage which accumulates in a complex stepwise, age-related fashion. Some tumours, however, result from a germ-line – usually inherited – gene mutation. This can give rise to a familial tumour predisposition syndrome ( Fig. 4.1 ), and the familial endocrine diseases discussed on the following pages are examples of such syndromes. They are typically characterised by predisposition to one or more tumours arising in endocrine and some neural crest-derived tissues, both benign (functional and non-functional endocrine tumours) and malignant (e.g. medullary thyroid cancer), and often separated by many years. Some individuals and families, however, only ever manifest with one tumour type: familial medullary thyroid cancer and familial hyperparathyroidism, for example.

The penetrance of an inherited disease is the proportion of individuals with the gene mutation (‘heterozygotes’, in the case of an autosomal dominant disorder) who develop disease. In the case of a multisystem disease, penetrance relates to any phenotypic manifestation. Note that penetrance of familial endocrine diseases is usually delayed until beyond childhood and is always less than 100%, hence the term ‘reduced penetrance’. This clearly implies that some individuals with a mutation in a familial endocrine disease gene will never develop disease. Such individuals may, of course, pass the condition to their offspring, who in turn may develop disease. Cascade testing that relies on clinical screening tests alone therefore has a false-negative rate; cascade testing using DNA testing is 100% sensitive, although it does not currently predict who will, and who will not, develop disease (see below).
Expression of an inherited disease is a description of the phenotypic manifestation. In the case of a single-phenotype disease, this may be ‘mild’ to ‘severe’, ‘unilateral’ or ‘bilateral’, etc.; in the case of a multisystem disease like multiple endocrine neoplasia type 1 (MEN1), this may be ‘pituitary adenoma plus primary hyperparathyroidism’, for example. Note that expression of familial endocrine diseases may change over time as an individual develops further disease manifestations.
Diagnostic genetic testing describes the process of identifying the disease-causing mutation in a DNA sample taken from the proband (the first affected family member to be identified). This may involve analysis of a single gene (e.g. in an individual with an MEN1 phenotype) or several genes (e.g. in an individual with an extra-adrenal phaeochromocytoma/ paraganglioma). There are three possible outcomes of a diagnostic test:
- •
Identification of a disease-causing mutation : a change in the gene sequence that has a predictable deleterious effect on gene function or protein chemistry and is therefore believed to be the cause of the proband’s disease.
- •
Identification of a variant of uncertain significance (VUS): a change in the gene sequence that usually results in an amino acid change in the corresponding protein but which has an unpredictable effect on that protein. In this situation, further investigations may clarify whether the variant is causally related to the phenotype. A VUS should never form the basis of a predictive genetic test (see below).
- •
No mutation or VUS identified: the mutation detection rate for a given gene in a particular clinical context is rarely 100%; in other words, the disease-causing mutation is undetectable in a proportion (usually small) of individuals with classical disease. Failure to identify a mutation has three possible implications:
- •
The diagnosis is correct but the gene mutation has not been identified. For example, a small proportion of probands with classical MEN1 have a mutation that cannot be detected using current technology (see under MEN1). This may also be a particular problem in diseases that can be caused by mutations in a number of different genes (e.g. familial paraganglioma/phaeochromocytoma), and should prompt the question ‘have we tested the correct gene?’
- •
The diagnosis is correct but the phenotype is not caused by a germ-line gene mutation, for example extra-adrenal paraganglioma/phaeochromocytoma.
- •
The diagnosis is not correct. For example, an 80-year-old woman with primary hyperparathyroidism, acromegaly and no family history of endocrine disease is likely to have a normal MEN1 genetic test result. The term phenocopy is used in this case to describe someone with coincidental ‘common’ endocrine problems that mimic MEN1.
- •
Cascade (‘predictive’) genetic testing describes the process of testing the proband’s relatives, once the disease-causing mutation has been identified. This should be done after full discussion about the consequences of a positive test result in terms of lifelong medical management, reproductive implications and life insurance. It is important to realise that cascade genetic testing simply identifies relatives who share the same mutation as the proband; it does not answer questions about penetrance or expressivity. In that sense, a cascade test can never be truly predictive.
Multiple endocrine neoplasia type 1 (MEN1)
MEN1 is an autosomal dominant familial syndrome characterised by the development of multiple and metachronous endocrine and non-endocrine tumours ( Table 4.1 ). Approximately 10% of cases arise de novo, without a prior family history of the syndrome. The precise prevalence of MEN1 is unclear. This in part refects variability in disease expression, even though penetrance may be high. The hallmark features of MEN1 are endocrine tumours of the pituitary, pancreas and parathyroid.
| Tumour/site * | Hormonal/other characteristics * |
|---|---|
| Parathyroid adenoma (90%) | |
| Enteropancreatic islet tumour (30–80%) | NF (80%)Gastrinoma (40%) Pancreatic polypeptidoma (20%) Insulinoma (10%) Glucagonoma VIPoma Somatostatinoma ACTHoma (rare) GRFoma (rare) |
| Anterior pituitary tumour (10–60%) | Prolactinoma (20%) NF (6%) GHoma (5%) ACTHoma (2%) |
| Foregut carcinoid | Gastric ECL tumour (10%) Thymic carcinoid (2–8%) Bronchial carcinoid (2%) |
| Adrenocortical tumour | Non-functioning adenoma (25%) Adrenocortical carcinoma (rare) Hyperaldosteronism (rare) |
| Cutaneous manifestations | Lipoma (30%) Angiofibroma (85%) Collagenoma (70%) |
* Values in brackets are estimates of penetrance of given characteristic at age 40.
Genetics
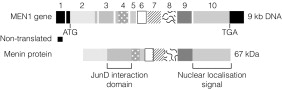
MEN1 is associated with heterozygous germ-line loss of function mutations in the MEN1 gene located on chromsome 11q13. Endocrine tumours from patients with MEN1 demonstrate loss of heterozygosity for the MEN1 locus, indicating that tumour formation is dependent on the development of a second somatic mutation in the wild-type allele ( Fig. 4.1 ). MEN1 therefore acts as a tumour suppressor gene. Heterozygous MEN1 mutant mice develop tumours mimicking the human phenotype. The MEN1 gene encodes a 67-kDa protein – menin – which has multiple functional domains ( Fig. 4.2 ). Menin can influence a number of key cellular processes including transcription, DNA repair and cytoskeletal function. Menin is known to bind several signalling proteins including JunD and Smad3. Recent data have highlighted menin’s role in the regulation of key developmental genes through influences on histone methylation.
Some patients and families manifest MEN1 but do not have demonstrable mutations in the MEN1 gene on gene sequencing. There are several potential explanations for this phenomenon:
- •
Mutation in a non-coding, regulatory region of the MEN1 (e.g. promoter).
- •
Presence of a whole exon deletion or duplication. Most mutation searching strategies now include an exon dosage assay.
- •
Disease mediated though an alternative MEN1 locus.
- •
Phenocopy – this refers to the chance ocurrence of two or more endocrine pathologies (both of which can be seen as part of MEN1) in the same person. One of these is usually primary hyperparathyroidism, which is a common sporadic condition, and the patient is usually over the age of 50.
The above possibilities should always be considered in an individual with possible MEN1 if MEN1 gene sequence analysis has been reported as normal.
MEN1 exhibits variable penetrance and variable expressivity (see above). Not all features of MEN1 will occur in a single patient or indeed a single family. Some families exhibit only hyperparathyroidism. There is considerable variation in age-related tumour penetrance and no clear genotype–phenotype correlation. It is therefore difficult to predict with any degree of accuracy the natural history of MEN1 in an individual or within a family.
Presentation
Presentation is dependent upon the herald lesion. More than one component may be apparent at presentation.
Primary hyperparathyroidism
Primary hyperparathyroidism (PHP) is the most common endocrinopathy in MEN1, and is thought to be present in at least 90% of cases aged 50 years or over. It is also the most common initial clinical expression of MEN1, with typical detection or presentation in the third decade of life, significantly earlier than that found in sporadic PHP. Patients with MEN1 generally have asymmetric, independent parathyroid adenomas in three or four glands. PHP often recurs following subtotal parathyroidectomy. PHP can exacerbate coexistent hypergastrinaemia from gastrinoma.
Enteropancreatic islet tumours
The prevalence of enteropancreatic islet tumours in patients with MEN1 may be as high as 80%, although the majority of such tumours are clinically silent and non-functional. Functional tumours can present in the second decade of life. Many a symptomatic patients have radiologically detectable tumours by the third decade. Tumours can arise throughout the pancreas and the duodenal submucosa. They are commonly multicentric, metachronous, and range in size and characteristics from micro- and macroadenomas to invasive and metastatic carcinoma. The prognosis of these tumours may relate to specific somatic molecular changes.
Up to 40% of patients with MEN1 develop gastrinoma, and current data suggest that up to 25% of all patients with gastrinoma have MEN1. Though presentation with invasive or metastatic disease is unusual before 30 years of age, metastatic disease (possibly occult) can be present in up to 50% of MEN1-associated gastrinoma at diagnosis. The presence of multiple, discrete gastrinomas can be mistaken for local disemminated disease. Tumours secreting pancreatic polypeptide are manifest biochemically and radiologically, but are generally clinically silent.
Pituitary tumours
The prevalence of pituitary tumours in MEN1 is uncertain, due to the range of patients and methods employed in the majority of studies to date. A large European multicentre study of 324 patients with MEN1 found pituitary tumours in 42% of cases. The most common pituitary lesion is prolactinoma. There are few prospective data on age-related penetrance of pituitary disease. However, MEN1-associated pituitary macroadenoma has occurred as early as 5 years of age.
Foregut carcinoids
MEN1-associated foregut carcinoid tumours are found in the thymus, stomach and bronchi. They are not generally hormonally active, and do not present with carcinoid syndrome. Their true prevalence is unclear. Gastric enterochromaffin-like (ECL) tumours are generally discovered at endoscopy. They exhibit loss of heterozygosity at the MEN1 gene locus and are promoted by hypergastrinaemia. Thus, they generally arise in MEN1 patients with gastrinoma. They can regress with normalisation of gastrin levels after surgical excision of gastrinoma. Thymic carcinoid disease has been highlighted as a major cause of mortality in MEN1. However, relatively little is known about its natural history. A prospective study of 85 patients with MEN1 found an incidence of 8% over a mean follow-up period of 8 years. Patients were all male, and most had no symptoms of the tumour at the time of detection. Interestingly, 4 of 7 of the tumours did not show somatic loss of heterozygosity at the MEN1 locus, raising questions as to the mechanism of tumour development. Serum chromogranin A was elevated in 6 of 7 tumours. Mean time interval between diagnosis of MEN1 and development of thymic carcinoid was 19 years. It may be that as early mortality reduces in MEN1 due to improved surgical and medical treatment, this relatively late expression of the disease increases in prevalence and impact.
Adrenocortical tumours
Adrenocortical disease occurs in 20–40% of patients with MEN1. It is unusual in patients who do not have pancreatic disease. Pathology may include diffuse hyperplasia, solitary adenoma and carcinoma. Disease can be bilateral. Excess hormone secretion is rare, and the majority of lesions are detected on routine radiological monitoring.
Cutaneous manifestations
A variety of cutaneous pathologies are now firmly established as components of MEN1. Cutaneous lipomas are often nodular and multicentric. Visceral lipomas have also been described. Cutaneous manifestations of MEN1 are useful clinically in the presymptomatic diagnosis of MEN1 in affected families.
Diagnosis
A diagnosis of MEN1 is considered in any patient presenting with two synchronous or metachronous tumours in the three characteristic sites (pituitary, pancreas and parathyroid). If there is a first-degree relative with a lesion typical of MEN1, the diagnosis should be considered in the presence of a single lesion. Patients with recurrent PHP, especially multiglandular disease, should have the diagnosis excluded.
The application of diagnostic DNA analysis has altered the phenotypic spectrum of MEN1, revealing both asymptomatic individuals and those with atypical phenotypes. DNA analysis does not always provide answers, however, as illustrated by phenocopies: the association of an endocrine tumour that has a low population prevalence – such as growth hormone (GH)-secreting pituitary tumour – with PHP could represent MEN1 or MEN1 phenocopy. Recent data suggest that mutations in the MEN1 coding region are infrequent in those patients without a family history of MEN1 who develop this combination of endocrinopathies. Absence of an MEN1 mutation may therefore be difficult to interpret, particularly if the patient is young and there is no supportive family history.
Management
MEN1 is associated with premature death, most commonly (30%) through metastatic islet cell tumours. Advances in the medical management of gastrinoma and hyperparathyroidism may result in a paradoxical increase in cumulative morbidity from other facets of the condition in the coming decade. The principal organs involved in MEN1 are difficult to screen for early tumours, and prophylactic surgery is either not appropriate or has not been shown to prevent the development of tumour (cervical thymectomy). The challenge is therefore to improve morbidity and mortality through targeted surgical and medical interventions as directed by surveillance and molecular screening programmes that aim to detect disease expression at an early stage in an inclusive manner.
Primary hyperparathyroidism
PHP in MEN1 is characterised by asynchronous involvement of all parathyroid glands. However, there remains debate as to the optimum type and timing of parathyroid surgery. Subtotal parathyroidectomy for PHP in MEN1 is associated with a surgical cure rate (as defined by the number of patients not hypercalcaemic) of 60% at 10 years and 51% at 15 years. The alternative, total parathyroidectomy with or without autograft, is associated with postoperative hypoparathyroidism and lifelong treatment with vitamin D analogues. Preoperative imaging and minimally invasive approaches may be difficult because of the need to examine all four glands. Transcervical thymectomy is recommended at the time of parathyroidectomy.
Enteropancreatic islet tumours
Enteropancreatic tumours in MEN1 are often multiple, recurrent and heterogeneous in behaviour. Correct management requires the correlation of symptoms, hormonal and imaging studies (which may be discordant), and experience in the natural history of the pathology. This can pose a significant challenge to the clinician.
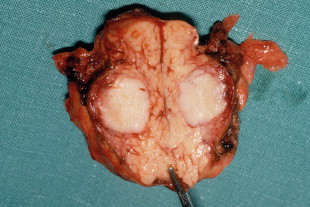
Surgery is the main treatment for patients with insulinoma in MEN1 ( Figs 4.3 and 4.4 ). All other syndromes of hormone excess due to enteropancreatic tumours respond well to medical therapy with proton-pump inhibitors (gastrinoma) or somatostatin analogues (VIPoma). The timing of surgery in the management of these conditions is debated.
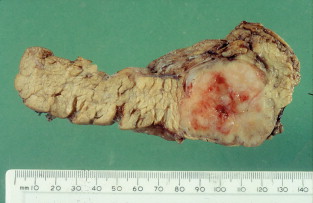
Gastrinomas in MEN1 are often multifocal and small, and can be situated in the duodenum. Extensive pancreatic–duodenal surgery can be associated with significant morbidity. Surgery for gastrinoma in MEN1 is frequently not curative, in part due to the multifocal nature of the problem. Furthermore, metastatic disease is found at surgery in a substantial number of patients in whom it is not apparent preoperatively. Nevertheless, the outcome of patients treated surgically for locally advanced disease can be the same as those with limited disease. Indeed, there are data that demonstrate that surgery is beneficial in increasing disease-related survival and decreasing advanced disease in Zollinger–Ellison syndrome.
A subset of MEN1 patients with gastrinoma has aggressive disease and decreased survival. Features associated with aggressive tumour behaviour include:
- •
diagnosis of MEN1 before 35 years of age;
- •
onset of gastrinoma at 27 years or younger;
- •
markedly elevated gastrin levels at presentation;
- •
tumour size greater than 3 cm.
Aggressive antitumour treatment in this group needs to be considered.
Non-functioning enteropancreatic islet cell tumours and those secreting pancreatic polypeptide are generally clinically silent. There is no consensus as to best treatment in this situation. Some advocate surgical removal if the lesion is greater than 3 cm or growing on serial radiological monitoring, while others suggest excision as a preventive measure in the absence of data suggestive of aggressive behaviour.
The standard surgical approach other than for gastrinoma is spleen-preserving distal pancreatectomy ( Fig. 4.5 ) and intraoperative bidigital palpation, coupled with intraoperative ultrasound and enucleation of any tumour found in the pancreatic head and duodenal submucosa. Surgery for gastrinoma should include duodenotomy. A Whipple procedure may be considered for tumours at the pancreatic head. Preoperative localisation of the target lesion with corroborative intraoperative ultrasound is useful in planning the appropriate approach. This can be important in the management of functional tumours as the pancreas and duodenum may contain multiple abnormalities, leading to uncertainty as to which of several lesions is the source of excess hormone production. Surgery prompted by abnormal biochemistry but in the absence of any scan-detected lesion should be considered to prevent malignant transformation of microadenomas. Distal 80% subtotal pancreatectomy should be considerd for risk modification in any paitent undergoing surgery for localised islet-cell tumour in MEN1.
Pituitary tumours
Pituitary tumours should be managed in the same manner as in isolated pituitary disease. Prolactinomas should be treated with dopamine agonists, with biochemical and radiological confirmation of response. Normalisation of prolactin levels without tumour shrinkage suggests misdiagnosis of a non-functioning pituitary adenoma with secondary hyperprolactinaemia. Non-functioning tumours should be treated with surgery. GH-secreting adenomas are best treated with primary surgery followed by consideration of external beam radiotherapy and somatostatin analogue therapy for persistent disease.
Foregut carcinoids
The optimum management of this generally late expression of MEN1 is unclear. Resection of bronchial carcinoid is usually required to make the diagnosis. Long-term follow-up is then required to check for recurrence. The natural history and malignant potential of ECL gastric carcinoids is unclear. Thymic carcinoid tumours are generally asymptomatic when detected through radiological screening, and can behave aggressively. Relapse is common after surgery, and the optimum adjuvant medical and radiotherapeutic approaches are not yet established.
Surveillance and screening
The multiple and metachronous nature of endocrine tumours associated with MEN1 requires lifelong clinical, biochemical and radiological surveillance to detect MEN1-associated tumour expression as soon as possible, minimising morbidity and optimising outcome. Genetic testing supports this process, facilitating the identification of both individuals within a kindred who will benefit from such long-term surveillance and those who do not require it.
Genetic testing
MEN1 gene analysis, involving sequencing of all coding exons of the MEN1 gene, should be offered to patients with MEN1 to help in determining biochemical and radiological screening strategies for their relatives. Analysis may also be helpful in those patients with atypical presentations, but only if an MEN1-defining mutation is found. Identification of an MEN1 mutation in an index case should lead to a screening cascade for the same mutation within the family, beginning with first-degree relatives. Given that 25% of all patients with gastrinoma have MEN1, genetic testing should be considered in patients presenting with gastrinoma, even in the absence of other features.
Absence of MEN1 coding region mutations does not necessarily exclude MEN1, and should trigger promoter and exon dosage studies if the index of suspicion of MEN1 is sufficient. If these prove negative in a family with suspected MEN1, linkage studies may be appropriate.
Biochemical and radiological surveillance
Biochemical and radiological screening should be offered to all patients with a diagnosis of MEN1, to asymptomatic relatives found to harbour an MEN1-defining MEN1 mutation on genetic testing, and to those found to be at risk through linkage studies ( Table 4.2 ). First-degree relatives of those patients with MEN1 in whom an MEN1 mutation has not been found should also be offered screening pending the outcome of promoter and exon dosage analyses. Biochemical and radiological screening should commence in early childhood, balancing age-dependent penetration, sensitivity of specific studies in specific age groups, and the inconvenience caused by the process. Screening should be lifelong for those patients with MEN1, those known to harbour MEN1-defining MEN1 mutations, and those defined as ‘at risk’ by haplotype and linkage studies. It should continue to the age of 50 in those kindreds in whom no genetic risk stratification is possible.
| Tumour type | Investigation | Age commencing (years) | Frequency |
|---|---|---|---|
| Parathyroid adenoma | iCa 2 + , PTH | 8 | Annual |
| Enteropancreatic islet cell | Gastrin | 20 | Annual |
| Glucose, insulin | 5 | Annual | |
| VIP, PP | 20 | Annual | |
| Glucagon | 20 | Annual | |
| Somatostatin | 20 | Annual | |
| MRI | 20 | 3- to 5-yearly | |
| Anterior pituitary | Prolactin | 5 | Annual |
| IGF-1 | Annual | ||
| MRI | 5-yearly | ||
| Foregut carcinoid | Chromogranin A | 20 | Annual |
| MRI | 20 | 3- to 5-yearly |
Gastrin levels are elevated in primary (atrophic) and secondary (drug-induced) achlorhydria, which can lead to false-positive screening tests for the disease. Ideally, treatment with H 2 antagonists and proton-pump inhibitors should be stopped for 2 and 4 weeks, respectively, before assessment of gastrin levels. However, gastrin levels in the normal range do not exclude gastrinoma, and there should be a low threshold for complementary corroborative gastric acid studies.
MEN1: differential diagnosis
Elements of MEN1 may rarely present as an isolated familial trait, or as part of a non-MEN1 syndrome. Enteropancreatic islet tumours and intestinal (foregut) carcinoid tumours usually occur either as sporadic tumours or as part of MEN1. Familial isolated enteropancreatic islet tumours have rarely, if ever, been described in the medical literature. Familial intestinal carcinoid tumours are extremely rare. Familial adenocortical diseases have been known for many years.
Familial isolated pituitary adenoma (FIPA)
Pituitary adenomas can occur in MEN1 and Carney syndrome (see below), as well as familial isolated pituitary adenoma (FIPA) syndrome. Indeed, familial acromegaly has been recognised for many years. FIPA is an autosomal dominant condition with variable penetrance: 15–25% of FIPA families harbour heterozygous mutations in the aryl hydrocarbon receptor-interacting ( AIP ) gene; in the remaining 80% of families the causative gene – or genes – remain(s) unknown.
Presentation
FIPA is characterised by early-onset pituitary adenoma, particularly somatotrophs, lactotrophs and somatolactotrophs. Corticotrophs, gonadotrophs and non-functioning pituitary adenomas (NFPAs) have also been described. Age of onset is younger than in sporadic pituitary adenomas, particularly so in families with AIP mutations. FIPA-related somatotroph adenomas appear to behave aggressively and response to somatostatin analogue therapy is poor.
Management
Identification of a mutation in the AIP gene not only serves to confirm the diagnosis but allows accurate cascade screening of the family. Mutation analysis of AIP is available in several laboratories worldwide. Individual features of the syndrome should be managed as in sporadic disease. Presentation of cortisol excess may be atypical and indolent. Diagnosis of FIPA syndrome should trigger periodic clinical, biochemical and radiological screening for additional features with the aim of reducing associated morbidity. However, there is currently no clear consensus as to the age this should commence, the method or the frequency of review/surveillance.
Familial intestinal carcinoid
This appears to be very rare. Germ-line sequence variants in the SDHD gene have been reported in a small series of apparently sporadic intestinal carcinoids, although it is not clear whether these represent disease-causing mutations. This may be akin to the identification of SDHD mutations in individuals presenting with apparently sporadic paraganglioma/ phaeochromocytoma (see below).
Multiple endocrine neoplasia type 2
MEN2 is an autosomal dominant familial cancer syndrome characterised by the metachronous development of medullary thyroid cancer (MTC), phaeochromocytoma and PHP. Overall penetrance of the disease is high in gene carriers although that of individual characteristics is varied. MEN2 is subclassified into several discrete forms with clinical, pathological and molecular correlates:
- •
MEN2A – MTC (90%), phaeochromocytoma (50%) and PHP (20–30%).
- •
MEN2A with cutaneous lichen amyloidosis.
- •
MEN2A with Hirschsprung’s disease (HD).
- •
Familial medullary thyroid cancer (FMTC) – at least 10 or more carriers or affected cases of MTC in a kindred over the age of 50 with no clinical or detectable evidence of other features of MEN2.
- •
FMTC with HD.
- •
MEN2B – MTC, phaeochromocytoma, decreased upper/lower body ratio, marfanoid habibitus, gastrointestinal and mucosal ganglioneuromatosis.
MEN2B is the most aggressive form, MTC presenting at a younger age and often with more advanced disease. Historically, the majority of MEN2B cases represent de novo mutations without a family history of the condition. Earlier diagnosis and improved management strategies may result in a change in this picture over the next 20 years.
Genetics
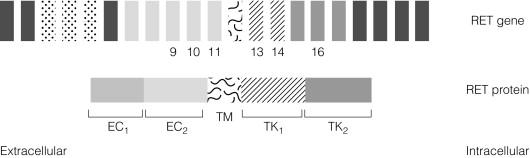
MEN2 is associated with heterozygous gain of function mutations in the RET gene found on Ch10q11.2. The RET gene codes for a membrane-associated tyrosine kinase with an extracellular cadherin-like domain and two independent intracellular tyrosine kinase (TK) domains ( Fig. 4.6 ). RET protein is expressed by a range of neuroendocrine cell types including the adrenal medulla, thyroid C-cells and parathyroid. In normal physiology, extracellular signals lead to RET dimerisation, triggering TK domain phosphorylation and a downstream signal transduction cascade leading to cell growth and differentiation. Gain of function mutations found in MEN2 produce constitutive activation of the RET signal transduction cascade outwith normal control processes.
MEN2A shows variable penetrance. Approximately 40% of gene carriers develop clinical manifestations by age 50 and 60% by age 70. Biochemical screening can lead to earlier identification of gene carriers: approximately 90% of individuals with MEN2A have biochemical abnormality by age 30 even if there are no overt signs of MEN2A.
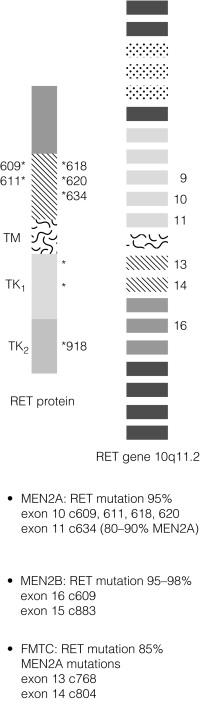
In contrast to MEN1, there is a partial genotype– phenotype correlation in MEN2. For the majority of families with MEN2A and FMTC the mutations in RET affect cysteine residues in the extracellular domain of the RET protein. The exact position of the cysteine residue involved by any particular mutation affects the likelihood of the phenotype being either MEN2A or FMTC. Virtually all mutations in MEN2A are found in exons 10 and 11 of the RET gene. For FMTC, mutations may be found in exons 13–15 as well as some in exons 10 and 11. For MEN2B, 95% have a mutation in exon 16 (codon 918), at a site that is prone to somatic mutation in sporadic MTC ( Fig. 4.7 ). There are data to suggest that there may be additional modifying factors, such as key RET single nucleotide polymorphisms (SNPs), that impact on disease expression within a given genotype. These may be particularly relevant in the situation of RET mutations that result in relatively weak constitutive activation.
Loss of function mutations in RET has been demonstrated in some kindreds with familial Hirschsprung’s disease. In contrast to the mutation hotspots noted in MEN2, these mutations are distributed throughout the gene.
Presentation
Presentation of MEN2A can be with any specific feature of the condition. MEN2B can present with additional signs or complications of ganglioneuromatosis (mucosal or gastrointestinal) prior to the development or recognition of an endocrinopathy. Some families present only with MTC.
Medullary thyroid cancer
MTC has been the first manifestation of MEN2 in most kindreds. It can present in the first decade of life with intrathyroidal, locally advanced or disseminated disease. Historically, MTC has been the major cause of morbidity and mortality in MEN2. Current management approaches will alter this natural history (see later). MTC in MEN2 is preceded by C-cell hyperplasia. Recent data have highlighted a 6.6-year window between development of MTC and progression to nodal metastases in MEN2A patients harbouring the most common (codon 634) RET mutation.
Phaeochromocytoma
Phaeochromocytoma can be unilateral or bilateral. Presentation can be with symptoms as in sporadic disease or as the result of positive surveillance studies. Phaeochromocytoma in MEN2 can present in the first decade of life.
Primary hyperparathyroidism
PHP occurs in 20–30% of patients with MEN2A, and is more common in those with RET codon 634 mutations. Most patients are asymptomatic. PHP associated with MEN2 is often less severe than that encountered in MEN1, and synchronous involvement of all four glands is less common.
Management
Medullary thyroid cancer
New cases of MEN2 presenting with MTC should be treated by thyroidectomy with central or more widespread node dissection, depending on pre- and perioperative staging. Thyroidectomy for MEN2B should include central node dissection. However, the aim of surgical management encompasses and is focused increasingly on prevention of MTC. Surgery for MTC in MEN2 should be performed before the age at which malignant progression occurs. Historically, this decision was based on basal and stimulated levels of the hormone calcitonin, produced by C-cells of the thyroid and a valuable tumour marker for MTC. However, this approach has an unacceptable sensitivity and specificity. Decisions on the timing of thyroidectomy in new cases of MEN2 without apparent MTC at presentation (such as those cases detected through genetic screening) should follow a stratified approach based on the genotype–phenotype relationships linking specific RET mutations with a specific natural history of MTC. Such an approach balances the earliest age at which MTC can present in association with a given RET genotype against the potential surgical morbidity of thyroidectomy at a young age ( Fig. 4.8 ).
Patients are assigned to one of three risk bands:
- •
Risk level 1 – all patients with MEN2B; patients with RET mutations involving codons 883, 918 and 922.
- •
Risk level 2 – patients with RET mutations involving codons 611, 618, 620 and 634.
- •
Risk level 3 – patients with RET mutations involving codons 609, 768, 790, 791, 804 and 891.
Patients in risk level 1 should undergo thyroidectomy in the first year of life, and preferably in the first 6 months of life. Those in risk level 2 should undergo thyroidectomy before the age of 5 years. There is no consensus on the optimum approach to patients in risk level 3. MTC presents at an older age in this group, and is commonly less aggressive. Recent data suggest that thyroidectomy need not take place before the age of 10 years, and that central node dissection is unnecessary before 20 years. The cumulative experience on which such recommendations are based remains limited.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


