Factor XIII (FXIII) is an unusual blood coagulation factor, circulating as a heterotetramer composed of two catalytic A-subunits and two noncatalytic B-subunits. It is a plasma transglutaminase (TG), mostly associated with fibrinogen, which is activated by thrombin in the presence of calcium in the final stages of the coagulation cascade. It acts by stabilizing the clot through the covalent cross-linking of polymerized fibrin chains and the incorporation of fibrinolysis inhibitors into the fibrin clot. Genetic polymorphisms in FXIII are associated with cardiovascular diseases (CVD), including coronary artery disease (CAD), stroke, and deep vein thrombosis (DVT). FXIII deficiency leads to severe bleeding, impaired wound healing, and recurrent miscarriages.
In this chapter, we review the basic functions of FXIII and focus on the most recent findings highlighting the role of FXIII in cardiovascular disease, in particular an emerging link to thrombosis. We use the detailed recommended FXIII nomenclature published in 2006 by the Scientific and Standardization sub-Committee of the International Society on Thrombosis and Haemostasis.1
DISCOVERY OF FACTOR XIII
The original discovery of a blood protein that makes fibrin chemically resistant to dissolution was made in the 1940s by two independent scientists. In the United States, Robbins2 described a serum factor that stabilizes fibrin during his PhD studies at the University of Illinois. At the same time, in Hungary and later in the UK, Lorand made similar discoveries of a blood protein involved in fibrin formation3,4 and discovered that the protein involved was from plasma rather than serum.5 Over the years, several names have been given to this factor including fibrinstabilizing factor, Laki-Lorand factor, fibrinoligase or fibrinase. During meetings of the International Society on Haemostasis and Thrombosis in the 1950s and early 1960s, it was decided that the name FXIII should be used, based on its main function, that is the stabilization of fibrin, and its position in the coagulation cascade.
FXIII was first purified from blood plasma by Lorand5 and later to further homogeneity by Loewy6,7,8 and Schwartz. It was discovered that FXIII is composed of two A-subunits and two B-subunits, arranged in a heterologous tetrameric structure.9,10,11 Discoveries of the first patient with FXIII deficiency and the fact that lack of FXIII leads to a serious bleeding disorder were made in Switzerland in 1960 by Duckert et al.12 The primary amino acid sequences for both the A- and B-subunits and the structure of the A-subunit gene have been elucidated by Ichinose and others,13,14,15,16,17,18 and, more recently, the crystal structure of the FXIII A-subunit (FXIII-A) dimer has been resolved at high resolution by Yee et al.19 and Weiss et al.20 These studies have paved the way for a profound understanding to be developed of the biology, structure, and function of this intriguing coagulation factor.
FACTOR XIII TETRAMER STRUCTURE
Cellular FXIII is composed of a dimer of A-subunits. FXIII in plasma is, however, composed of two A- and two B-subunits9,10 (FIGURE 17.1). Binding to the B-subunits is necessary to provide stability to the hydrophobic A-subunit in the aqueous environment of the blood; without FXIII B-subunit (FXIII-B) the A-subunit is unstable as demonstrated by the absence of FXIII-A in blood plasma in patients with congenital B-subunit defi-ciency21 The structure of FXIII-A2B2 is that of a heterologous tetramer, with a molecular weight of 325.8 kDa.22 FXIII-A forms a dimer with a central axis of symmetry, and the two monomers aligned in opposite N- to C-terminal orientation as characterized by x-ray crystallography19,20 (FIGURES 17.1 and 17.2). The tertiary structure of FXIII-B and that of the tetramer remains, however, unknown. The closest information we get on the structure of the FXIII-A2B2 tetramer is that obtained by rotaryshadowed FXIII analyzed by electron microscopy (EM).23 This study showed that FXIII-A2 demonstrates a globular structure, whereas FXIII-B in isolation is a monomer and demonstrates a flexible strand-like structure. It has been suggested that the two B-subunits wrap around the globular A-subunit dimer in a protecting manner.23 However, the resolution is too low to conclude with confidence on the structure for the FXIII-A2B2 tetramer, and future studies of higher resolution will be needed to determine the structural requirements for interaction between the A- and B-subunits. FIGURE 17.1 shows a schematic representation of the FXIII-A2B2 tetramer for illustration purposes; the conformation of FXIII-B in this structure in particular is hypothetical.
FXIII-A is produced as a typical cytoplasmic protein by monocytes, macrophages, and megakaryocytes.24,25,26 The protein has no leader sequence for the classical secretion pathway.14,17 Recent evidence suggests the existence of nonclassical secretion mechanisms for FXIII-A from macrophages.27 FXIII-B on the other hand is synthesized in the liver and secreted via the classical secretion pathway from hepatocytes.28 At which site the FXIII-A2B2 tetramer is assembled, and by which mechanism, remains unknown. As FXIII-B is normally secreted and circulates in excess, one line of thought is that FXIII-B could be delivered to the site of synthesis for FXIII-A and that it might be required to aid the release of FXIII-A from the cell. Phenotype changes for FXIII-A after bone marrow transplantation, and for FXIII-B after liver transplantation, suggest that these two organs are the main sites of synthesis for the FXIII subunits.29
FIGURE 17.1 Schematic representation of F×lll A2B2 tetrameric structure.Two F×lll-A molecules (blue) are aligned in opposite N- to C-terminal orientation around a central axis of symmetry. The flexible, strand-like B-subunits (green) are thought to wrap around the hydrophobic A-subunits. The conformation of the B-subunits in this representation is hypothetical and based on previous EM studies.23 The F×lll activation peptides (APs) are depicted in red.
FACTOR XIIIA-SUBUNIT, CATALYTIC TRIAD, AND CALCIUM BINDING
The gene for the FXIII-A is located on chromosome 6 bands p24-25.30,31 It contains 15 exons that code for a 731-amino acid mature protein.17 The FXIII-A gene shows significant sequence homology with those of the other TGs, in particular keratinocyte TG 1 and tissue TG 2.32,33,34 However, while most TG genes are <30 to 40 kb in size, the FXIII-A gene spans more than 160 kb, due to the large size of the introns in this gene. The FXIII-A gene shows linkage with other genes on chromosome 6 including the human leukocyte antigen genes of the major histocompatibility complex31 and the plasminogen gene at bands q26-27.35
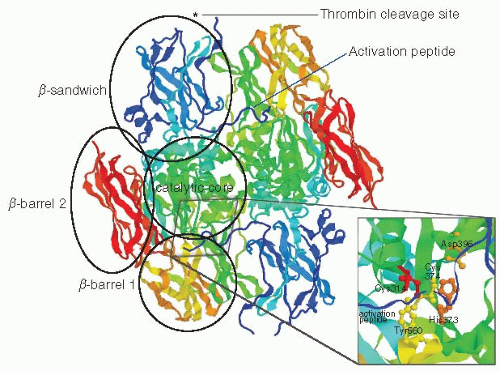
FIGURE 17.2 Tertiary structure of the F×XIII A2 dimer. F×XIII-A forms a dimer of two subunits in antiparallel orientation. Each A-subunit is composed of (from N- to C-terminus) an AP that is cleaved by thrombin at peptide bond Arg37-Gly38 (indicated by an asterisk, not resolved on the crystal structure), a β-sandwich domain, a catalytic core, a β-barrel 1 domain, and a β-barrel 2 domain. The inset shows a zoom in on the catalytic core, with the catalytic triad, Cys314, His373, and Asp396 highlighted in red, orange, and ocher, respectively. Residues Tyr560 from the β-barrel 1 domain and residue Cys374, which protect the active site Cys314 from oxidation, are highlighted in yellow. Image was drawn using RasMol 2.7.5 and the crystal structure coordinates from Weiss MS, Metzner HJ, Hilgenfeld R. Two non-proline cis peptide bonds may be important for factor XIII function. FEBSLett 1998;423(3):291-296 (PDB file 1F13).
The mature FXIII-A protein has a molecular weight of 83.2 kDa22 and is composed of five structurally and functionally distinct regions, which from N- to C-terminus include an activation peptide (AP) (residues 1 to 37), a yβ-sandwich domain (38 to 184), the catalytic core (185 to 515), a β-barrel domain 1 (516 to 628), and a β-barrel domain 2 (629 to 727)19 (FIGURE 17.2). The catalytic triad involved in transamidation is composed of Cys314, His373, and Asp396.19 FXIII-A2 is a dimer of two subunits in antiparallel orientation. The AP from one subunit crosses the interface and interacts with the second subunit (FIGURES 17.1 and 17.2). FXIII-A shares its basic domain structure with many other members of the TG enzyme family. However, most other TGs lack an AP and do not form a dimer.36,37 It has been suggested that dimerization of FXIII-A enhances substrate specificity of this enzyme by increasing the surface available for substrate binding.36
FXIII-A contains nine cysteine residues, none of which are disulfide bonded. Oxidation of the active site Cys314 leads to loss of enzyme activity. In the catalytic core of the zymogen, Cys314 is sandwiched between another cysteine residue (374) and an aromatic tyrosine residue (560) to protect it from oxidation.19,20 Tyr560 is located in β-barrel domain 1, and hence removal of this residue from the active site by a conformational change of β-barrel 1 could play an important role in the exposure of Cys314 for interaction with the substrate. A conformational change that leads to β-barrel 1 separating from the catalytic core is exactly what happens when tissue TG is activated,37 and it is therefore possible that a similar conformational change is involved in FXIII activation. Evidence for large conformational changes during the activation of FXIII-A is still scant. An x-ray crystal structure of activated FXIII-A did not show any major conformational changes compared with FXIII-A zymogen.38 However, studies of solvent accessibility of residues in FXIII-A by chemical labeling of surface exposed cysteines and lysines did indicate that conformational changes of FXIII-A are likely to play an important role in activation.39 Feasible conformational changes in FXIII-A during activation have recently been modeled based on the x-ray crystal structure of active tissue TG.40
Calcium plays important roles both in FXIII activation and in the catalytic mechanism (see below). The main calcium binding site in FXIII-A is composed of residues Asn436,Asp438,Ala457, Glu485, and Glu490 of the catalytic core region.41 Mutations at Glu485 and Glu490 significantly reduced catalytic activity of activated FXIII (FXIIIa),in agreement with a major role of these residues in calcium binding.42 This calcium binding site can be found at the dimer interface and is located very close (<10 Å) to β-barrel domain 1. Its location close to β-barrel domain 1 suggests that calcium binding may be involved in the conformational change that removes yβ-barrel domain 1 and Tyr560 away from the catalytic core during activation.41 Location of the calcium binding site at the dimer interface may also account for the allosteric effects of calcium binding on the exposure of the catalytic site for substrate interaction in both subunits, even if only one of them has been cleaved by thrombin, as reported by Hornyak and Shafer.43
Another structurally important characteristic of FXIII-A, which could be involved in conformational changes of the catalytic domain during activation, involves the presence of two nonproline cis-peptide bonds in FXIII-A at Arg310-Tyr311 and Gln425-Phe426, an uncommon feature in protein structures.20 These nonproline cis-bonds, together with the Gly410-Pro411 cis-bond, could be involved in a major conformational “switch” in the catalytic core when it is transformed into an energetically favorable transconformation.20 In agreement with this, mutations in either Arg310 or Tyr311 have been shown to result in an inactive FXIII molecule, indicating the potential importance of one of the two nonproline cis-bonds in FXIII activation.44
FACTOR XIII B-SUBUNIT STRUCTURE AND FUNCTION
The gene for FXIII-B is located on chromosome 1 bands q32-32.1 and is 28 kb in size.45 It contains 12 exons separated by 11 introns and codes for a 641-amino acid mature protein.46 The FXIII-B gene is part of a cluster of homologous genes located at bands q31-32 on chromosome 1, including the genes of five complement regulating proteins, factor H,47,48 C4 binding protein,49 C3b/C4b receptor,50 decay accelerating factor,51 and membrane cofactor protein.52 The products of the genes at this locus contain a varying number of short consensus repeats called Sushi domains.15,53,54 Sushi domains are typically involved in protein-protein interactions, and the FXIII-B protein is entirely composed of 10 Sushi domains with a 20-amino acid leader sequence for secretion and a short 11-amino acid C-terminal sequence.15 Each Sushi domain contains four cysteine residues that participate in two disulfide bonds that provide stability to the structure of the domain. In total, there are therefore 20 disulfide bonds in FXIII-B. The tertiary structure of FXIII-B is hitherto unknown; however, FXIII-B appears as a long filament, or as “beads on a string,” when imaged by EM.23 Using recombinant FXIII-B and truncations of the molecule, it has been suggested that the first Sushi domain is responsible for binding FXIII-A and that the fourth and ninth Sushi domains are responsible for binding to another B-subunit.55
The B-subunit plays a number of roles in FXIII physiology. First, two B-subunits bind two A-subunits to form the heterologous tetramer that provides stability to the protransglutaminase in blood plasma. Whether FXIII-B also somehow aids the secretion of FXIII-Afrom its source of production is unknown, but it has been suggested that some hepatoma cell lines (PLC/PRF/5) are capable of expressing both FXIII-A and FXIII-B and that they secrete intact FXIII-A2B2.56 Second, the dissociation of the B-subunit from FXIII-A is a critical event in the activation of FXIII (see below). FXIII-B may also play an important role in the interaction of FXIII with fibrinogen γ (see below), thereby directing the cross-linking activity of FXIIIa to one of its major substrates. It is not known if FXIII-B is involved in other protein interactions.
FACTOR XIII ACTIVATION
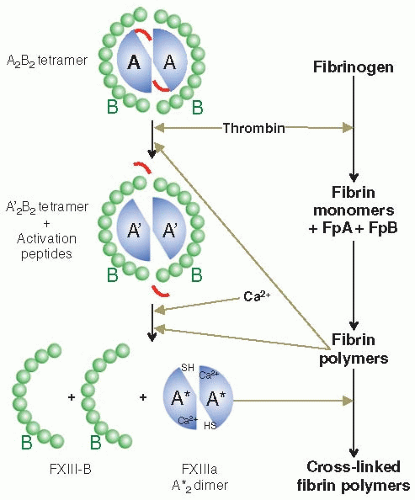
FXIII-A2B2 is converted into its active form by a two-stage process (FIGURE 17.3). The first step involves the cleavage of the Arg37-Gly38 peptide bond by thrombin that results in the release of an N-terminal AP of 37 amino acids to generate FXIII-A, followed by the second step of calcium-dependent dissociation of the B-subunits from the cleaved A-subunits. The cleaved A′-subunits in the absence of the B-subunits adopt an active conformation in the presence of calcium, FXIIIa.57,58 As a consequence of FXIII activation, the active site cysteine, which is usually buried and inaccessible, is unmasked allowing access to potential substrates. Under physiologic circumstances thrombin is not able to cleave FXIII without the presence of the cofactor, fibrin.59 Fibrin has been shown to enhance both steps of FXIII activation (FIGURE 17.3). Polymeric fibrin I and II (des-A- and des-A, B-fibrinogen) enhance the rate of thrombin cleavage of the A-subunits by approximately 80-fold. Fibrin I and II reduce the Michaelis constant (Km) for thrombin during the activation of FXIII by forming a ternary complex with thrombin and FXIII, enabling activation to occur under physiologic conditions of clot formation.59,60,61 The fibrin enhancement effect on thrombin cleavage only occurs with FXIII-A2B2 and not cellular FXIII-A2.62,63 Fibrin also accelerates the rate of A- and B-subunit dissociation following thrombin cleavage and it has been reported that the α-chains of fibrin play a role in this.64 Calcium is essential for enabling the dissociation of FXIII-A from FXIII-B by modulating the conformation of FXIII-A.39,65 Fibrin enhancement of FXIII activation ensures that the formation of FXIIIa only occurs at the site of a forming fibrin clot, where FXIIIa crosslinking activity is required.
FIGURE 17.3 Activation of FXIII. F×XIII is activated in a two-step process: in a first step thrombin cleaves the AP from FXIII-A, and in a second step the B-subunits dissociate from the active A-subunit dimer in the presence of calcium. Both steps are enhanced by the presence of the main substrate of F×XIIIa in plasma, fibrin. Polymerizing fibrin increases cleavage of FXIII-A by thrombin as well as subunit dissociation. Fibrin formation and F×XIII activation are therefore closely regulated to occur at the same time.
A second thrombin cleavage site (Lys513-Ser514) has been identified in the FXIII-A16 This site is not considered to play an important role in vivo because its cleavage is only observed in vitro when thrombin is incubated with FXIII in the absence of calcium, but not in its presence.66
TRANSGLUTAMINASE REACTION
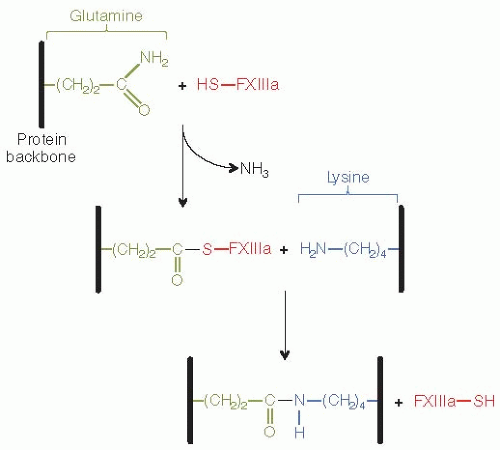
Unlike the other eight members of the TG family mainly found in tissues, FXIIIa is a plasma-specific endo-γ-glutamine:ε-lysine transferase enzyme that plays an important role in the formation of fibrin gels.7 It catalyzes protein cross-linking reactions by specifically replacing the ammonia of a glutamine residue67,68 with an amino group from a lysine residue,69,70 therefore creating a covalent isopeptide bond between these two amino acids. Upon activation by thrombin, FXIIIa cross-links adjacent fibrin molecules and induces stabilization of the fibrin clot.
The first step of the most common fibrin γ-chain crosslinking process is the reaction of the thiol group of factor XIIIa active site Cys31471 with the carbonyl of the γ-glutamine residues (398/399) of a fibrin molecule (Table 17.1, FIGURE 17.4), resulting in the generation of an acyl enzyme intermediate and release of ammonia (FIGURE 17.5). The second step requires the recognition of a closely located Lys406 from an adjacent fibrin molecule (Table 17.1) aligned during fibrin polymerization, and the subsequent formation of an isopeptide bond between the ε-amino group of the lysine and the γ-glutamine residue, resulting in the cross-linking of γ-chain fibrin and the release of FXIIIa (FIGURE 17.5). If the appropriate lysine substrate is not available, the glutamine residue can then be deaminated (Fibrin-(CH2)2-CO-S-FXIII + H20 ↔ FXIII-SH + Fibrin-(CH2)2-COOH), or linked to a substrate primary amine, the latter resulting in a peptide bond (Fibrin-(CH2)2-CO-S-FXIII + H2N-Amine FXIII-SH + Fibrin-(CH2)2-CONH-Amine).72
The amino acid sequences surrounding both glutamine and lysine residues play some role in substrate recognition. This peptide sequence specificity is particularly important for recognition of glutamine by FXIIIa. Studies have shown that FXIIIa only recognizes specific glutamine residues,73 and that the peptide sequence surrounding this amino acid plays a role in substrate recognition.74 The latter study, using amino acid substitution, highlighted the requirement of leucine (pos-4), lysine (pos+2), valine (pos+3), and leucine (pos+4) residues in the vicinity in order for the glutamine residue (pos0) to be recognized by FXIIIa. The nature of the amino acids preceding the lysine residue is less influential on the cross-linking reaction. A comprehensive study has shown that on one hand, valine, arginine, and phenylalanine have a favorable effect to substrate reactivity, with serine, alanine, leucine, tyrosine, and asparagine also promoting cross-linking. On the other hand, glycine, aspartic acid, or to a lesser extent proline, histidine, and tryptophane have an adverse effect on creating an isopeptide bond between lysine and glutamine.75 It is possible that tertiary structure surrounding the glutamine and lysine residues plays a more important role in substrate recognition for FXIIIa than primary structure. Future structural studies on the interaction of FXIIIa with different substrates will be required to elucidate this further.
Although fibrin γ-chain dimerization is the major product of FXIIIa enzymatic activity, fibrin α-chains cross-linking putatively occurs at Gln221, 237, 328, and 366, and Lys208, 219, 224, 418, 427, 429, 448, 508, 539, 556, 580, 601, and 606 (Table 17.1, FIGURE 17.4) at a much slower rate and results in highly cross-linked α-chain multimers.76,77,78 Fibrin α-γhybrid heterodimers and multimers79 and γ-chain trimers/tetramers80 have also been documented but are normally not a major product of FXIIIainduced cross-linking.
To date, no human isopeptidase has been identified that is capable of disrupting the covalent ε(γ-glutamyl)lysyl bond created during fibrin cross-linking, but FXIIIa itself has been reported to display effective isopeptidase activity in vitro, with Km values of 10ˆ-4-5.81 However, the physiologic relevance of this reaction and the mechanisms by which it occurs remain unclear.
HOMOLOGYWITH OTHER TRANSGLUTAMINASES
TGs are a family of ubiquitous enzymes, with presence in bacteria, plants, and animals. They are part of a superfamily of enzymes that are classified as R-glutaminyl-peptide:amine γ-glutamyltransferases (EC 2.3.2.13).82 The reactions catalyzed by the EC 2.3.2.13 enzyme family include transamidation or cross-linking, deamidation, and amine incorporation.83 The biologic functions of TGs span a plethora of different activities, from involvement in egg fertilization, semen coagulation, cell death, and epidermal and hair structure to blood coagulation.
In humans, besides FXIII, there are at least eight other TGs (Table 17.2). These include keratinocyte TG or TGI involved in epidermis formation,84 tissue TG or TG2, a ubiquitous enzyme involved in a variety of cell functions including cell death,85,86,87 TG3 involved in hair follicle stabilization,88 TG4 with unknown function in the human prostate,89 TG5 involved in epidermal differentiation,90 TG6 and TG7 with unknown functions,91 and erythrocyte protein 4.2 that plays a role in erythrocyte membrane integrity92 The genes of the TG family demonstrate significant sequence homology, and the proteins show remarkable structural similarity with a basic domain structure that includes a yβ-sandwich domain, and a catalytic core followed by two β-barrel domains. The catalytic core regions of TGs are significantly conserved, with a 75% sequence homology between the active site of TG2 and FXIII-A for example.82 The only member of the TG superfamily that does not contain a functional active site is erythrocyte protein 4.2.93 This protein, although not active as a TG, is very abundant in the human red blood cell membrane where it plays a significant role in membrane stability through interactions with CD47, band three, and ankyrin, as demonstrated by hereditary spherocytosis (a form of hemolytic anemia) caused by protein 4.2 deficiency94
Clot retraction, stabilization of the platelet cytoskeleton
α,IIb/β226,228,230
—
Fibrin
Stabilization of the platelet—fibrin clot
αVβ229,230
—
—
Adhesion of platelets to endothelial cells
a Gln2 and Gln14 relate to the same glutamine residue from the short and long forms of ¢2-AP respectively.
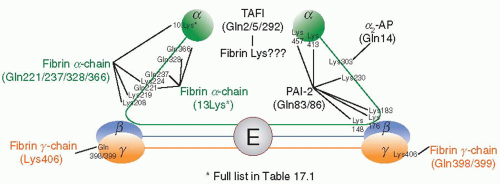
FIGURE 17.4 Fibrin cross-linking sites. Schematic of the fibrinogen molecule, with the central E-region, coiled coil region, and two distal D-regions.The sites where F×XIIIa introduces cross-links are indicated in the fibrinogen molecule.The γ-chain (orange) of fibrin is cross-linked at residues Lys406 and either Gln398 or 399. The α-chain (green) has known cross-linking sites for fibrinolysis inhibitors such as α2-AP and PAI-2, and many sites have been reported to be involved in αcross-linking.
FIGURE 17.5 Transglutaminase reaction by F×XIII. F×XIIIa active Cys314 reacts with a substrate glutamine to generate an acyl enzyme intermediate and release a molecule of ammonia.The acyl enzyme then reacts with a lysine substrate, resulting in the formation of covalent ε(γ-glutamyl)lysyl bond and release of F×XIIIa.
FXIII has several unusual characteristics compared with other TGs. Whereas FXIII-A (the TG part of the proenzyme) forms a heterologous tetramer with FXIII-B, most other TGs exist in the form of a monomer. The only other TG that is not a monomer is rat TG4 that has been reported to form a homodimer, similar to FXIII-A.83 Another uncommon feature of FXIII is that its activation involves proteolytic cleavage by thrombin in the presence of calcium. Most other TGs are activated by the presence of calcium alone, but there are two other exceptions to this. TG3 is activated by proteolytic cleavage by dispase that leads to a 50 kDa N-terminal fragment containing the catalytic core and a 27 kDa C-terminal fragment.95,96 The activation of TGI is also regulated by an additional mechanism, but unlike FXIII-A and TG3 not by limited proteolysis. Activation of TGI in the absence of extracellular calcium is regulated by conformational changes induced by interaction with tazarotene-induced gene 3 (TIG3).97,98 TIG3 is also a substrate for TGI, hence it is interesting to compare its stimulatory effect on TGI activation with the enhancing effect that fibrin has on FXIII activation (see above).
Most TGs are intracellular, cytoplasmic proteins, apart from FXIII-A and TG2 both of which are secreted. TGI is anchored to the cell membrane via palmitate and myristate linkages at a cluster of five cysteine residues in the N-terminus of the protein.99 TG2 demonstrates guanosine triphosphate (GTP) hydrolizing activity in addition to its cross-linking activity100,101 This GTPase activity competes with cross-linking activity in TG2, as binding of GTP inhibits the cross-linking function of TG2.102 Cross-linking by FXIIIa is however unaffected by GTP,102 indicating that FXIII does not support GTPase activity.
NTERACTIONS WITH FIBRIN(OGEN) AND ROLE OF FIBRIN α-CHAIN
As mentioned above, the fibrin α-chain contains several potential cross-linking sites, and its cross-linking by FXIIIa leads to high molecular weight polymers. All the α-chain cross-linking sites are located in the C-terminal half of the chain (αC), which contains many protein binding sites and plays an important role in lateral aggregation of fibrin.103 The cross-linking of this chain by FXIIIa therefore contributes to the stability of the laterally aggregated fibrin fiber.
Several studies have been performed to elucidate the interactions of various forms of FXIII with fibrinogen and fibrin. Greenberg’s group showed that nonactivated FXIII-A2B2 bound specifically to fibrinogen via the FXIII-A2 subunits with an equilibrium constant (KD) of 10 nM104 and identified that FXIII-A2B2 binding also occurred with fibrinogen polymerized with protamine sulfate, fibrin I produced with reptilase and fibrin II, indicating that binding of FXIII is not dependent on thrombin or the release of fibrinopeptides.105 The authors went on to propose a model wherein FXIII zymogen reversibly binds to fibrinogen (via the A-subunits), remaining there after cleavage of fibrinopeptide A, which is cleaved from the fibrinogen α-chain by thrombin during the conversion of fibrinogen to fibrin. At this point the AP of the A-subunits of FXIII is cleaved by thrombin during the formation of fibrin II polymers. Once the A-subunits of FXIII are cleaved, the B-subunits are able to dissociate in the presence of calcium to form FXIIIa.105 Hornyak and Shafer63 compared thrombin-cleaved, nonactivated platelet FXIII-A and nonactivated FXIII-A2B2 for binding to fibrin. The thrombin-cleaved FXIII-A2bound with a higher affinity to fibrin (KD = 2.1 µM) compared to noncleaved FXIII-A subunit (KD =14 µM). However, the tetramer FXIII zymogen (A2B2) bound to fibrin with a much greater affinity (KD = 0.2 µM) than either form of the A-subunit preparations. These authors saturated fibrin with plasma FXIII zymogen, and this did not modify the interaction of thrombin-cleaved FXIII-A subunit, suggesting FXIII zymogen and FXIIIa bind to separate sites on fibrin.
Lorand’s group established that residues 242 to 424 of the fibrin α-chain play a role in enhancing the dissociation of the A- and B-subunits of FXIII-A2B2 following thrombin cleavage.64 This work therefore indirectly infers that these residues of the α-chain are involved in the binding of thrombin-cleaved FXIII-A2B2, which is likely to involve the A-subunit. Subsequent work by Greenberg’s group106 showed, using immunoblotting techniques, that binding of FXIII-A took place on the carboxy-termini of the α- and yÖ-chains, but not on the E fragment (which is the central fragment of the molecule that contains the N-termini of all the chains) of fibrinogen (see FIGURE 17.4 for diagram of fibrinogen structure). Procyk et al.107 found that thrombin-activated FXIII-A subunit bound to residues 389 to 402 of the α-chain, by competing the binding of FXIIIa to fibrin columns with antibodies directed against this sequence of the fibrin α-chain. More recently, taking a direct approach of recombinant expression of truncated fibrinogen α-chains and using surface plasmon resonance to study the interaction of FXIIIa to these α-chain fragments, FXIII-A was found to bind to residues 233 to 425 in the αC with a K of 2.4 µand this interaction was further localized to aC residues 389 to 403. Using a site-directed mutagenesis approach, it was found that mutation of the single residue Glu396 of the αC- chain abolished the binding to FXIIIa.108
The fibrin γ-chain is one of the preferred substrates for FXIIIa; its cross-linking is rapidly completed (5 to 10 minutes) after clot formation. FXIIIa cross-links the γ-chains between Lys406 and one of two tandem glutamine residues situated at position 398 and 399.109,110 Cross-linking occurs when two γ-chains are lined up, the D-regions (which contain the C-terminal domains of both the γ- and the Ö-chains of fibrin) facing each other, in the half-staggered D-E-D fibrin protofibril arrangement of fibrin (see FIGURE 17.4 for diagram of fibrinogen structure). Two γ-chain cross-links are introduced in this assembly across the D-dimer interface in an antiparallel orientation.111
In agreement with the structural orientation of the γ-chain at the D-D interface, fibrin γ-chain cross-linking does not normally lead to higher molecular weight complexes, such as those formed during cross-linking of the α-chains, but leads to the formation of γ-chain dimers.112 When cross-linked fibrin is digested by plasmin, proteolysis of the coiled coils on either side of the cross-linked D-regions leads to the formation of D-dimers.113,114,115 Mutation of Lys406 in the fibrinogen γ-chain modifies the topology of fibrin cross-linking by FXIIIa with the production of γ-a hybrid cross-links.116 Similar γ-α hybrid cross-links are also found when fibrin is cross-linked by TG2,117,118 suggesting that Lys406 is a specific acyl-acceptor for FXIIIa and not for TG2.
Some studies have suggested that the γ-chain cross-links introduced by FXIIIa show a different configuration than the longitudinal orientation of antiparallel cross-links across the D-D interface. Mosesson suggested that γ-chain cross-links show a transverse orientation, spanning from one γ-chain in one protofibril across to a second in another protofibril, suggesting that this agrees with fiber extensibility upon stretching of the fiber, which has lead to some debate on this topic in the literature.119,120 Recent data from atomic force microscopy studies of fibrin fibers at stretch, however, has provided further evidence for the γ-chain longitudinal cross-linking model121 in agreement with previous x-ray crystallography122 and EM studies.123
Cross-linking of the fibrin γ-chains is inhibited by a monoclonal antibody against the C-terminal region of the γ-chain124 and by clumping factor A from Staphylococcus aureus that binds to the same region.125 Residue Ile387 of the fibrin γ-chain seems to be important for the interaction of the γ-chain with FXIIIa, as mutation of this amino acid impairs γ-chain cross-linking.126
It has been suggested that FXIIIa binds to the fibrinogen γ-chain C-terminal region. In particular, a splice variant of the γ-chain, fibrinogen γ, has been reported to interact with FXIIIa. Studies using zymogen FXIII-A2B2 suggested that the γ-chain of fibrinogen was responsible for binding FXIII-A2B2, as FXIII-A2B2 copurifies with *γγ but not γhlγh fibrinogen. Contrary to this work, studies by Lord’s group, using recombinant expression of γhlγh and γγ fibrinogen, found similar binding affinities for FXIII-A2B2, regardless of the presence of γ127 Fibrinogen γ is caused by alternative polyadenylation,128 which leads to two messengers differing at the C-terminus. For the γ-chain, the last 4 residues are replaced by 20 different residues.129 FXIII copurifies with the fibrinogen γ-chain, and this effect is due to the B-subunit, as A-subunit on its own does not coelute.130,131 These findings suggest that FXIII-B may bind to the γ sequence, which is negatively charged and also contains a thrombin-binding site.132,133,134 FXIII also binds to a γ peptide, which competitively inhibits binding of FXIII to fibrinogen.135 Fibrinogen γ has been shown to associate with increased activation of FXIII.136 The mechanisms behind the *ggr;/FXIII interaction remain largely unknown and further studies will be needed to characterize the physiologic relevance of these findings.
ROSS-LINKING OF INHIBITORS OF FIBRINOLYSIS
FXIIIa cross-links several protease inhibitors to fibrin, in order to protect the clot from breakdown (fibrinolysis) (Table 17.1, FIGURE 17.4). Plasmin is the main protease responsible for fibrinolysis, and its major inhibitor is the serine protease inhibitor α2-antiplasmin (α2-AP). Deficiencies and abnormalities in plasma α2-AP result in severe hemorrhagic disorders,137,138,139,140,141,142,143,144,145,146 characterized by increased susceptibility to fibrinolysis, which can be reverted in vitro by addition of purified α2-AP to plasma from deficient patients.147 FXIIIa is able to cross-link Gln2/14,21,419, 447 of α2-AP148,149,150 to Lys303 of α-chain fibrin.151 Circulating free α2-AP inhibits tissue plasminogen activator (tPA)-induced fibrinogenolysis,152 whereas FXIIIa-mediated cross-linking of α2-AP to fibrin results in inhibition of fibrinolysis.153 Plasmin bound to fibrin is protected from inhibition by α2-AP.154,155 There are two forms of α2-AP circulating in human plasma.
Only gold members can continue reading. Log In or Register to continue

 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Molecular Basis for Platelet Secretion
Molecular Basis for Platelet Secretion
 Acquired Nonimmune Thrombocytopenia
Acquired Nonimmune Thrombocytopenia
 Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient



