Factor XI
Peter N. Walsh
David Gailani
Coagulation factor XI (FXI), previously known as plasma thromboplastin antecedent, was first identified by Rosenthal et al.1 as a deficiency in the plasma of patients with abnormal bleeding and, subsequently, was found to be particularly common in patients of Ashkenazi Jewish descent.2,3 FXI was first purified from bovine4 and human5 plasma as a 160-kDa homodimeric protein that participates in interactions with coagulation proteins of the contact phase of blood coagulation.6,7,8,9 Factor XI is the zymogen of a trypsin-like serine protease that is activated by FXIIa and by both thrombin and FXIa,10,11 and it can then recognize FIX as its normal macromolecular substrate in plasma.12,13,14
GENE STRUCTURE AND REGULATION
The approximately 23 kb human FXI gene contains 15 exons and is located on the distal end of the long arm of chromosome 4 (4q35).15,16 The FXI cDNA derived from human liver mRNA is 2,097 nucleotides long.17 The first exon is untranslated, while the second encodes the signal peptide. The factor XI polypeptide in plasma contains 607 amino acids encoded by exons 3 to 15 (see FIGURE 15A.1).15,17,18 Exons 3 to 10 encode four repeats called apple domains (A1 to A4 from the N-terminus).18 The intron-exon boundaries are located at similar positions within each apple domain, indicating duplication from a common ancestral sequence.15 Apple domains are members of the PAN (Plasminogen, Apple, Nematode) module family, with homology to the N-terminal domains of hepatocyte growth factor and plasminogen19 and similar core topology to PAN domains from leech antiplatelet protein,20 the protozoan microneme antigen EtMIC5,21 and apical membrane antigen 1 from Plasmodium falcipurum.22
Exons 11 to 15 of the FXI gene encode the trypsin-like catalytic domain and are organized similarly to genes for tissue plasminogen activator and urokinase.15 The proteases thrombin and factors VIIa, IXa, and Xa have tyrosine or phenylalanine in their catalytic domains at the position corresponding to residue 225 in chymotrypsin. This confers Na+-induced allosteric regulation on the catalytic domain and is associated with an AGY codon for the active site serine residue (Ser195, chymotrypsin numbering), consistent with these proteases having evolved from a common thrombin-like protease.23,24 In contrast, FXIa and the contact proteases α-kallikrein and FXIIa have proline at position 225, indicating they are not regulated by sodium. Pro225 is associated with a TCN codon for Ser195, suggesting these proteases evolved from a trypsin-like protease distinct from the ancestor of the vitamin K-dependent proteases.23
The amino acid sequence of human FXI is 58% identical to plasma prekallikrein (PK, the zymogen of α-kallikrein),17 which also consists of four apple domains and a trypsin-like catalytic domain.18,25 The PK gene contains 15 exons, with intron-exon boundaries in identical positions to those of the FXI gene,26 indicating the genes are products of a duplication event. Consistent with this, exon 15 of the PK gene is only approximately 8 kb from exon 1 of the FXI gene in humans.27 Analyses of vertebrate genomes reveal that fish do not have homologs for FXI or PK.28,29 A gene encoding a serine protease with four apple domains that is clearly ancestral to both proteins is found in amphibians, birds, reptiles, and the primitive egg-laying mammal (monotreme) the duck-billed platypus.29,30 The proteins encoded by the ancestral genes have features that are more similar to PK than FXI.30 Distinct genes for FXI and PK are present in the opossum, a marsupial, and in placental mammals, indicating duplication of the ancestral gene occurred after the common lineage leading to these two groups diverged from monotremes.30
The normal site of synthesis of plasma FXI is the liver. FXI levels decrease in liver disease, and patients have acquired,31,32 or been cured of,33,34 FXI deficiency by liver transplantation. Northern blot analysis confirms that the liver is the major, if not exclusive, site of FXI mRNA synthesis in mice.35 In humans, FXI mRNA has been found by northern blot35 and reverse-transcriptase polymerase chain reaction (RT-PCR)36 in liver, pancreas, and kidney; and by RT-PCR in platelets.37,38,39 Immunohistochemistry studies confirmed that FXI protein is present in human hepatocytes, pancreatic islet cells, and the proximal and distal tubules in the kidney.40 The sequence between base pairs -385 and -350 of the human FXI gene promoter contains a binding site for hepatocyte nuclear factor-4α (HNF-4α), a transcription factor that appears to be required for hepatocyte-specific FXI expression.27 Mice with a liver-specific deletion of HNF-4α have reduced plasma FXI levels.41 HNF-4α is expressed at high levels in human pancreatic islets and kidney,42,43 explaining the presence of FXI in these tissues. There is a small amount of FXI activity associated with platelets (˜0.5% of the activity in plasma),44,45,46,47,48 but it is not clear that FXI mRNA found in platelets is responsible for this. Platelet FXI is discussed in detail below.
Large epidemiologic studies have demonstrated an association between plasma FXI level and venous49 and arterial thrombotic disease50; however, genetic factors controlling FXI gene expression are not well understood. Common single nucleotide polymorphisms (SNPs) are present in the human FXI gene promoter at base pair -403 (G/T) and -273 (C/G), the latter of which clearly alters transcription factor binding in gel mobility shift assays.51 However, it is not clear that there is a relationship between these SNPs and plasma FXI level. Two common SNPs within the FXI gene have recently been associated with deep
venous thrombosis (DVT) and high plasma levels of FXI.52,53 Interestingly, the SNPs remained associated with risk of DVT even after adjustment for FXI level,53 but the mechanisms by which these polymorphisms contribute to thrombosis risk is not known.
venous thrombosis (DVT) and high plasma levels of FXI.52,53 Interestingly, the SNPs remained associated with risk of DVT even after adjustment for FXI level,53 but the mechanisms by which these polymorphisms contribute to thrombosis risk is not known.
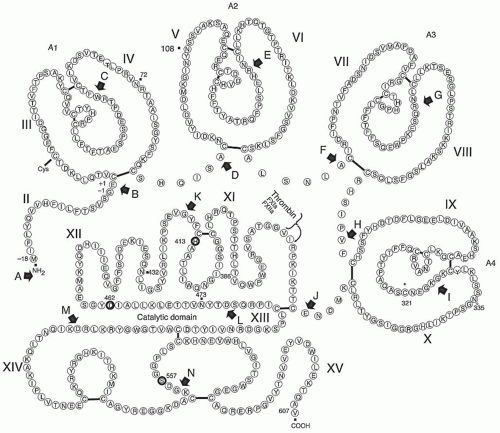 FIGURE 15A.1 Amino acid sequence and primary structure of human FXI. The signal sequence (numbered -18 to -1) is removed during biosynthesis by signal peptidase by the cleavage of the G-E bond between -1 and -2. Factor XI circulates in plasma as a homodimer connected by a single disulfide bond linking C321 in both of the fourth apple domains. The 14 introns (A to N) are shown by solid arrows. The exons are denoted by II to XV amino- to carboxy-terminus, with exon II representing the propeptide and exon I (not shown) representing the 5′ untranslated region. The four apple domains (consisting of 90 to 91 amino acids) are labeled as A1, A2, A3, and A4. The site of cleavage catalyzed by thrombin, factor XIa, or factor XIIa during the conversion of factor XI to factor XIa is shown with a small, curved arrow. The three members of the catalytic triad (H413, D462, and S557) are circled in bold. The four N-linked carbohydrate chains (i.e., N72, N108, N432, and N473) are shown by solid squares. (Figure redrawn and modified from McMullen BA, Fujikawa K, Davie EW. Location of the disulfide bonds in human coagulation factor XI: the presence of tandem apple domains. Biochemistry 1991;30:2056-2060.) |
Full-length and partial FXI cDNA sequences are now available for more than a dozen mammalian species. The best characterized of these are the murine35 and rabbit54 cDNAs, which encode proteins of 624 amino acids (18 amino signal peptides and 606 amino acid mature polypeptides) with 78% and 87% amino acid sequence homology to human FXI. In most species, the two polypeptides of the FXI homodimer are covalently linked by a disulfide bond involving Cys321 in the A4 domain.55,56 The single known exception is rabbit FXI, which migrates as a monomer during denaturing gel electrophoresis due to histidine replacing Cys321.54 Western blot analysis using a monoclonal antibody that recognizes a conserved epitope on the apple 2 (A2) domain confirms that FXI from species
other than the rabbit can form disulfide bond-linked dimers.57 Despite lacking the Cys321-Cys321 bond, rabbit FXI exists in plasma as a noncovalently associated homodimer that is functionally identical to human FXI.54
other than the rabbit can form disulfide bond-linked dimers.57 Despite lacking the Cys321-Cys321 bond, rabbit FXI exists in plasma as a noncovalently associated homodimer that is functionally identical to human FXI.54
There is little detailed information regarding intracellular processing and posttranslational modification of FXI. The protein undergoes N-linked glycosylation at Asn72, Asn108, Asn432, and Asn473, but not at the potential glycosylation site at Asn335.17 Dimer formation is an intracellular event that was initially thought to be required for normal secretion of the protein, based on observations that naturally occurring mutations in the fourth apple domain that interfere with dimerization are associated with poor secretion.58,59 However, it is now apparent that some forms of monomeric FXI with mutations that disrupt the dimer interface are synthesized and secreted similarly to wild-type protein in culture.56 Failure to secrete some FXI variants that have defects in dimer formation, therefore, are likely related to structural perturbations distinct from the defect in dimerization.
PROTEIN STRUCTURE
Human FXI (Mr ˜ 143,000) is a disulfide-linked homodimer containing approximately 5% carbohydrate (i.e., 0.6% hexose, 2.7% N-acetylhexosamine, and 1.7% N-acetylneuraminic acid), and is present in human plasma at a concentration of 4 to 6 µg/mL (˜30 nM) in the form of a zymogen that requires proteolytic activation to develop serine protease activity.5,8,60,61,62 The zymogen form of the protein is unique among coagulation factors because it is a disulfide-linked homodimer consisting of two identical polypeptide chains. Each FXI polypeptide contains 607 amino acids, and each can be proteolytically activated by FXIIa, thrombin, or FXIa5,8,9,10,11 at an internal R369-I370 bond to yield a heavy chain (i.e., 369 amino acids) and a light chain (i.e., 238 amino acids) that contain a typical trypsin-like catalytic triad, consisting of H413, D464, and S557, with a typical trypsin-like substrate-binding pocket.17,18 After cleavage at R369-I360 bond, the catalytic domain contains an amino-terminal sequence of I-V-G-G characteristic of most serine proteases. The Mr of the homodimer is 135,979 without carbohydrate, and 143,000 with four carbohydrate chains at N72 and N108 within the heavy chain and at N432 and N473 within the catalytic domain, whereas no carbohydrate is present on N335, another potential N-glycosylation site.17
The primary sequence and domain structure of FXI, including its disulfide linkages, are shown in FIGURE 15A.1. The amino-terminal 369 residues of each FXI monomer after propeptide cleavage comprises the heavy-chain region of FXIa, which consists of four tandem repeat sequences (Apple domains) of 90 to 91 amino acids, each containing six or seven cysteine residues that form three internal disulfide bonds. The amino acid sequences of each apple domain are 23% to 34% identical to the other apple domains of FXI and 58% identical to the corresponding apple domains of plasma PK. In both plasma FXI17,18 and plasma PK, a characteristic disulfide-bonding pattern links the first and sixth, second and fifth, and third and fourth cysteines in each of the four apple domains.18 An additional cysteine is present at position 11 in the A1 domain of FXI and forms a disulfide bond with another cysteine residue, whereas C321 in the A4 domain is disulfide-linked to the corresponding C321 to form the homodimer.18
Crystal structures have recently been reported for full-length FXI63 (see FIGURE 15A.2), and for the catalytic domain of FXIa in complex with both synthetic and natural inhibitors including
the Kunitz protease inhibitor (KPI) domain of protease nexin-2 (PN2)64,65,66. The structural features of FXI and their implications for molecular function have recently been discussed in detail67. In addition, the solution structure of the FXI A4 domain has been reported68 that predicts a major conformational change in the structure of FXI when it is activated to FXIa.
the Kunitz protease inhibitor (KPI) domain of protease nexin-2 (PN2)64,65,66. The structural features of FXI and their implications for molecular function have recently been discussed in detail67. In addition, the solution structure of the FXI A4 domain has been reported68 that predicts a major conformational change in the structure of FXI when it is activated to FXIa.
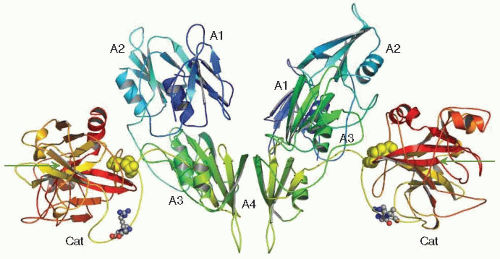 FIGURE 15A.2 X-ray crystal structure of human factor XI zymogen. The FXI dimer is shown with the two monomeric subunits interacting with noncovalent interactions mediated by residues within the two Apple 4 (A4) domains and stabilized by a disulfide bond (not shown) between the two Cys321 residues in each of the two A4 domains. The four apple domains constituting the heavy chain region of each monomer are designated A1, A2, A3, and A4 and the catalytic domains (light chains of FXIa) are designated Cat. The disulfide bonds between Cys362 in the A4 domain and Cys482 in the catalytic domain are designated by the yellow space-filling ball structures. The site (Arg369-Ile370) cleaved by FXIIa, thrombin, or FXIa is designated by the small space-filling balls in the connecting loops between the A4 domains and the catalytic domains. The fine arrows point at the active site Ser557 residues within each of the catalytic domains. (Figure modified from Papagrigoriou E, McEwan P, Walsh PN, et al. Crystal structure of the factor XI zymogen reveals a pathway for transactivation. Nat Struct Mol Biol 2006;13:557-558.) |
PLATELET FACTOR XI
There are differing opinions regarding the possibility that human platelets contain FXI activity and that the activity is associated with a form of FXI encoded by an alternatively spliced mRNA from the FXI gene. FXI coagulant activity and antigen have been described in washed platelet suspensions, and it has been estimated that the activity constitutes up to 0.5% of the FXI activity in normal plasma.44,45,46,47,69 Polyclonal, but not monoclonal, antibodies to human plasma FXI cross-react with a platelet protein with an apparent Mr of 220,000 on nonreducing SDS-PAGE which decreases to 55,000 after reduction. For comparison, plasma FXI has an Mr of 160,000, and each subunit has an Mr of 80,000.44,45,46 Thus, it has been suggested that platelet FXI could represent a disulfide-linked tetramer of identical 55,000-Mr subunits. Alternatively, the 55,000-Mr protein could be disulfide linked to a platelet membrane protein.37,45,70 A candidate for this putative platelet FXI-binding protein is glycoprotein Ib (GP Ib) (Mr ˜ 170,000). This possibility is supported by studies demonstrating that platelets from patients with Bernard-Soulier syndrome who lack GP Ib contained no detectable FXI activity in washed platelet suspensions.71 It is important to point out, however, that the 55,000-Mr species has not been shown to have FXI activity (i.e. the capacity to activate factor IX) in functional assays.
If the 55,000-Mr platelet protein is encoded by an mRNA transcribed from the FXI gene, the mRNA would need to be spliced differently than FXI mRNA in hepatocytes. FXI mRNA is present in platelet RNA; however, there are conflicting reports on its nucleotide sequence.37,38,39,72 The FXI mRNA in hepatocytes is 2.1 kb in size. A 1.9 kb mRNA transcript was isolated from a megakaryocytic leukemia cell line (MEG-01) that had an identical open reading frame to the hepatocyte species, with the exception that it lacked nucleotides corresponding to exon V.37 This would result in deletion of amino acids A91 through R144 within the A2 domain. By contrast, two other groups have reported that the FXI mRNA in platelets and megakaryocyte cell lines (including MEG-01) is identical in sequence to FXI mRNA in liver.38,39 Furthermore, attempts to express a FXI cDNA lacking exon V in a fibroblast line resulted in a poorly expressed protein of 73,000-Mr that was not secreted.73 Thus, the data linking the platelet protein detected with the polyclonal anti-FXI antibody or the FXI mRNA species detected in platelet mRNA to FXI activity are not compelling, and it is not currently possible to conclude that the FXI activity associated with platelets is distinct from plasma FXI of hepatocyte origin.
STRUCTURE-FUNCTION RELATIONSHIPS
Factor XI Activation
Plasma FXI circulates as a zymogen in human plasma as a complex with high molecular weight kininogen (HK),7 which is required for binding of FXI to negatively charged surfaces74 (see Chapter 15B). FXI can be activated by four biologically relevant proteases: FXIIa, FXIa, thrombin, and meizothrombin.5,10,11,75,76 It can participate in the contact phase of blood coagulation in a reaction that requires the presence of anionic surfaces for optimal surface-mediated in vitro activation by FXIIa.6,7,74,77,78 However, because deficiencies of FXII, PK, and HK are not associated with hemostatic abnormalities but deficiency of FXI produces abnormal bleeding complications in at least 50% of affected individuals,2,3,79,80,81 it has been suggested that the more relevant in vivo pathway for activation of plasma FXI might be by the feedback activation by thrombin or, possibly, by autoactivation by FXIa.10,11,82 It has been shown that β2-glycoprotein I (β2-GPI) binds FXI and inhibits its activation by thrombin and FXIIa, whereas cleaved β2-GPI binds FXI but fails to inhibit FXI activation.83 All four proteases (i.e., thrombin, FXIIa, FXIa, and meizothrombin) cleave each monomer of FXI at the R369-I370 bond, generating the new amino-terminal sequence of the catalytic domain, I-V-G-G, which then activates the catalytic triad of the serine protease. It has been postulated that on anionic surfaces, such as in extracorporeal circulation, a ternary complex is formed when FXI, in stoichiometric complex with HK in plasma, is adsorbed to the negatively charged surface, where FXIIa recognizes it as a substrate.6,7,74,77,78,84,85,86,87 The FXI homodimer is thereby cleaved to generate two disulfide-linked heavy chains (Mr ˜ 50,000) and two active-site-containing light chains (Mr ˜ 30,000).5,8 Recent observations demonstrate that FXI activation by thrombin or FXIIa proceeds through the generation of an intermediate consisting of one cleaved and one uncleaved subunit, referred to as 1/2-FXIa, which can be detected in plasma undergoing contact activation.88
An important role for the dimeric structure of FXI in zymogen activation has been demonstrated. Thus, the A4 domain of FXI mediates dimer formation between the two identical polypeptide chains comprising FXI through disulfide bond formation at C321,17,18,55,56,58,89,90,91 and the two identical subunits of FXI utilize a substantial interface (886 Å2) between two adjacent A4 domains to form the dimeric structure of the mature protein.63,68 Utilizing FXI with substitutions for Cys321, which still form stable dimers, it has been demonstrated that hydrophobic interactions involving Leu284, Ile290, and Tyr329, and salt bridges between Lys331 of one subunit and Glu287 of the other56 are required for dimer formation, and that FXI dimer formation is required for efficient FXI activation by thrombin, FXIIa, and FXIa.55,56,63,68,89,90,91,92
Functional Domains of Factor XI
Prior to the availability of FXI structures from x-ray crystallography and NMR and based on the use of molecular models of the four FXI apple domains,93,94,95,96,97 monoclonal antibodies, conformationally constrained synthetic peptides, and recombinant proteins98 with site-directed mutations, the binding sites within FXI for a number of physiologically relevant plasma or cellular ligands have been identified and characterized.
Thrombin,99,100 HK,73,93,95,101 and the kringle 2 domain of prothrombin103 have all been shown to bind to contiguous subdomains within the carboxy-terminal half of the A1 domain (E1-S90). Thus, HK binds to a subdomain within the A1 domain comprising residues F56-S86, whereas thrombin binds to a partially overlapping and contiguous site comprising subdomain A45-R70. Different amino acid side chains appear to be essential for these interactions because E51 and E66 are important for thrombin interactions with FXI but not for HK binding, whereas V64 and I77 are important for HK binding but not
for thrombin binding101. The kringle 2 domain of prothrombin can bind to a subdomain comprising residues A45-S86, but fine mapping of this site has not yet been accomplished.102 Occupancy of these sites by their respective ligands is likely to be important in promoting the binding of FXI the GP Ibα subunit of the GP Ib/IX/V complex on the platelet surface100 either in the presence of HK and Zn2+ ions or prothrombin and Ca2+ ions.
for thrombin binding101. The kringle 2 domain of prothrombin can bind to a subdomain comprising residues A45-S86, but fine mapping of this site has not yet been accomplished.102 Occupancy of these sites by their respective ligands is likely to be important in promoting the binding of FXI the GP Ibα subunit of the GP Ib/IX/V complex on the platelet surface100 either in the presence of HK and Zn2+ ions or prothrombin and Ca2+ ions.
The A3 domain of FXI has been identified as the locus of binding sites for both platelets97,103,104,105,106,107,108 and heparin.109,110 Thus, activated platelets expose specific reversible, high-affinity binding sites for FXI that require the presence of HK and ZnCl2,111 or prothrombin and CaCl2.102 Binding of FXI to activated platelets through the A3 domain97,111 is required for optimal rates of FXI activation by FXIIa.112 The sequence of residues within the A3 domain of FXI (i.e., N235-R266) that comprises a contact surface for interaction with a platelet receptor consisting of the GP Ibα subunit of the GP Ib/IX/V complex107,108 contains amino acids that mediate this binding, including N248, R250, K255, F260, and Q263.104,106 The A3 domain of FXI also contains all the binding energy required for interaction of FXI with heparin in a heparin-binding consensus sequence (i.e., T249-F260) that contains amino acids K252 and K253, and possibly K255, which at least in part mediate the binding of FXI to heparin.109,110
A major role of the A4 domain of FXI is to mediate dimer formation, the functional significance of which is discussed in detail above in the section on FXI activation. Furthermore, a natural mutation in FXI (F283L), resulting in diminished intracellular dimerization and secretion of the protein,58 also markedly affects dimer dissociation and the unfolding mechanism of FXI, indicating that F283 is important for noncovalent dimer formation.91 In addition, a sequence of residues (i.e., A317-G350) within the A4 domain of FXI appears to contain three peptide structures, possibly consisting of three antiparallel β-strands that together comprise a contact surface for interaction with FXIIa. This site may participate in the activation of FXI by FXIIa.96,103
CELLULAR INTERACTIONS
Early observations suggesting that platelets participate in the contact phase of blood coagulation113,114,115,116,117 were subsequently confirmed by the evidence that isolated human platelets activated with either adenosine diphosphate or collagen could promote the proteolytic activation of FXII by kallikrein and, subsequently, the activation of FXI by FXIIa in the presence of HK.47,112,118,119 The mechanism underlying the role of activated platelets in the activation of FXI involves the generation of high-affinity (dissociation constant, Kd, ˜10 nM), specific, saturable binding sites (˜1,500 sites per platelet) on the surface of activated platelets that require the presence of HK and Zn2+ ions.111 The observation that HK also binds to platelets under similar conditions originally suggested that the two proteins bind to the platelet surface as a complex.120,121 However, it has subsequently been shown that HK interacts with FXI through a surface-exposed site encompassing residues F56-S86 within the A1 domain93,95,122, resulting in the exposure of a platelet binding site encompassing residues N235-R266 within the A3 domain of FXI.97 It has also been demonstrated that prothrombin can substitute for HK as a cofactor for the binding of FXI to the surface of activated platelets.102 In this way, prothrombin appears to bind to a site in the A1 domain of factor XI contiguous to the site used by HK for binding to the A1 domain.102 The platelet receptor that binds factor XI and colocalizes it with thrombin for efficient activation has recently been shown to consist of the GP Ibα subunit of the GP Ib/IX/V complex that is localized within platelet membrane microdomains referred to as lipid rafts.107,108 This interaction of FXI with surface membranes of activated platelets has been shown to be mediated by binding of residues (i.e., R250, K252, K253, K255, F260, and Q263) within the A3 domain of FXI104 to the leucine-rich repeat motifs of GP Ibα.123 The binding of FXI to activated platelets and activation by thrombin are mediated by the interaction of FXI and platelet GP Ibα with thrombin anion-binding exosites I and II, respectively.100,124,125
Related posts:
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Molecular Basis for Platelet Secretion
Molecular Basis for Platelet Secretion
 Acquired Nonimmune Thrombocytopenia
Acquired Nonimmune Thrombocytopenia
 Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
Pathogenesis and Treatment of Biomaterial-Associated Thrombosis
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


