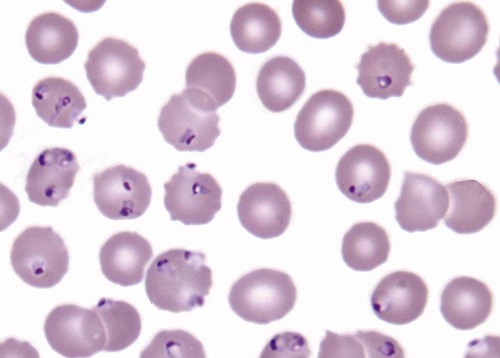
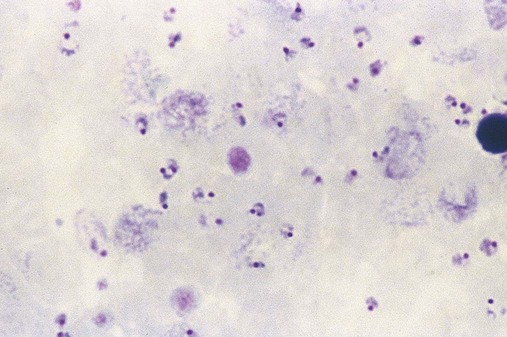
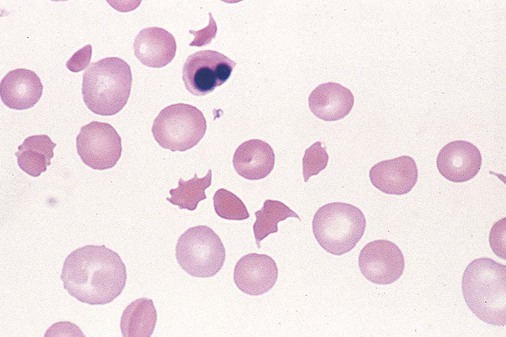
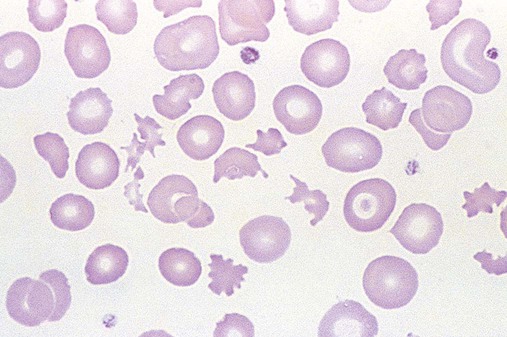
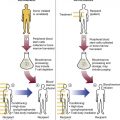
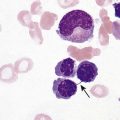
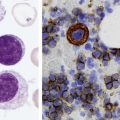
After completion of this chapter, the reader will be able to: 1. Describe the general pathophysiology and clinical laboratory findings in microangiopathic hemolytic anemia, including the characteristic red blood cell morphology. 2. Compare and contrast the pathophysiology, clinical symptoms, and typical laboratory findings in thrombotic thrombocytopenic purpura, hemolytic uremic syndrome, HELLP (hemolysis, elevated liver enzymes, and low platelet count) syndrome, and disseminated intravascular coagulation. 3. Explain the pathophysiology and typical laboratory features of hypertensive crises, traumatic cardiac hemolytic anemia, and exercise-induced hemoglobinuria. 4. Describe the life cycle of Plasmodium, including the hepatic and erythrocytic cycles, and the insect vector. 5. Explain the pathophysiologic mechanisms in P. falciparum infection that lead to anemia and neurologic manifestations. 6. Differentiate the five Plasmodium species affecting humans based on the geographic distribution, characteristic morphology on a peripheral blood film, the extent of parasitemia, and the length of the erythrocytic cycle. 7. Describe the proper specimen collection and procedure for performing a thin and thick blood film examination. 8. Compare and contrast Babesia species and Plasmodium species in terms of geographic distribution, clinical symptoms of infection, and morphology. 9. Describe the pathophysiology, laboratory findings, and peripheral blood morphology in hemolytic anemia due to clostridial sepsis, bartonellosis, drugs, chemicals, venoms, and extensive burns. 10. Given the history, symptoms, laboratory findings, and a representative microscopic field from a peripheral blood film of a patient with suspected extrinsic, nonimmune hemolytic anemia, discuss possible causes of the anemia and indicate the data that support the conclusions. Inclusions were noted on the thin and thick peripheral blood films (Figures 24-1 and 24-2). 1. Identify the inclusions present on the thin and thick peripheral blood films. 2. What is the likely diagnosis for this patient? 3. What clues in the history support this diagnosis? 4. What other forms might be found on the peripheral blood films in this disease? 5. What are the pathophysiologic mechanisms for the anemia in this disease? Extrinsic hemolytic anemias comprise a diverse group of disorders in which red blood cells (RBCs) are structurally and functionally normal, but a condition outside of the RBCs causes premature hemolysis. The extrinsic hemolytic anemias can be divided into conditions with nonimmune and immune causes. A common feature in the nonimmune extrinsic hemolytic anemias is the presence of a condition that causes physical or mechanical injury to the RBCs. This injury can be caused by abnormalities in the microvasculature (microangiopathic) or the heart and large blood vessels (macroangiopathic), infectious agents, chemicals, drugs, venoms, or extensive burns. The nonimmune disorders causing hemolytic anemia are discussed in this chapter and are summarized in Box 24-1. In disorders in the immune category, the hemolytic anemia is caused by antibodies, complement, or both, and these conditions are covered in Chapter 25. Examination of a peripheral blood film is important in suspected extrinsic hemolytic anemias, because observation of abnormal RBC morphology, such as schistocytes, spherocytes, or the presence of intracellular organisms, provides an important clue to the diagnosis. Microangiopathic hemolytic anemias (MAHAs) are a group of disorders characterized by RBC fragmentation, hemolysis, and anemia. The fragmentation occurs intravascularly by the mechanical shearing of RBC membranes as the cells rapidly pass through turbulent areas of small vessels that are partially blocked by microthrombi or damaged endothelium.1,2 Upon shearing, RBC membranes quickly reseal with minimal escape of hemoglobin (Hb), but the resulting fragments (called schistocytes) are distorted and become rigid.1 The spleen clears the rigid RBC fragments from the circulation through the extravascular hemolytic process (see Chapter 22).1 Laboratory evidence of the hemolytic anemia includes a decreased hemoglobin and hematocrit (Hct), reticulocytosis, increased serum unconjugated bilirubin, decreased serum haptoglobin, and increased urine urobilinogen. In some cases, the fragmentation is so severe that intravascular hemolysis occurs with varying amounts of hemoglobinemia, hemoglobinuria, and markedly decreased levels of serum haptoglobin.1 The presence of schistocytes on the peripheral blood film is a characteristic feature of microangiopathic hemolytic anemia. The RBC shearing may also produce helmet cells and, occasionally, microspherocytes. Polychromasia and nucleated RBCs are found when the anemia is severe. Thrombotic microangiopathy is a more specific term used to describe disorders in which the microangiopathic hemolytic anemia is due to the formation of thrombi in the microvasculature. In addition to the fragmentation hemolysis, thrombocytopenia and ischemic organ injury are characteristic findings. The thrombotic microangiopathies include thrombotic thrombocytopenic purpura (TTP), hemolytic uremic syndrome (HUS), and HELLP (hemolysis, elevated liver enzymes, low platelet count) syndrome.3 Disseminated intravascular coagulation (DIC) can also be classified as a thrombotic microangiopathy; however, schistocytes are found in the peripheral blood film in only about half of the cases.4 TTP and HUS may be difficult to differentiate, because they can have overlapping clinical and laboratory findings (Box 24-2); however, they have different etiologies and require different treatments.5 This chapter provides a summary of these conditions. TTP, HUS, and HELLP syndrome are discussed in more detail in Chapter 43. DIC is discussed in more detail in Chapter 42. TTP is a rare disorder characterized by the abrupt appearance of microangiopathic hemolytic anemia, severe thrombocytopenia, and a markedly elevated level of serum lactate dehydrogenase.2,6 Neurologic dysfunction, fever, and renal failure may also occur, but they are not consistently present.2 TTP is more commonly found in adults and is rare in children, and there is a higher incidence in females than in males.6,7 Typical laboratory findings at presentation include a hemoglobin concentration of less than 10 g/dL, a platelet count of less than 20 × 109/L, and schistocytes observed on the peripheral blood film (Figure 24-3). After the bone marrow begins to respond to the anemia, polychromasia and nucleated RBCs may also be observed. The white blood cell (WBC) count is often increased, and immature granulocytes (left shift) may appear. The bone marrow shows erythroid hyperplasia and a normal number of megakaryocytes. Various amounts of protein, RBCs, and urinary casts may be present in the urine depending on the extent of the renal damage. Results of coagulation tests are usually within the reference range, which differentiates TTP from DIC. The pathophysiology of TTP involves the release of long von Willebrand factor (VWF) multimers from vascular endothelial cells.8 The long VWF multimers adhere to endothelial cells and rapidly bind platelets through specific receptor-ligand interaction and subsequently trigger platelet aggregation. In the process platelets are removed from circulation, which causes severe thrombocytopenia, and the platelet-VWF (hyaline) microthrombi block small blood vessels, which leads to ischemia in the brain, kidney, and other organs.2,8 In addition, RBCs are sheared as they pass through small vessels partially blocked by microthrombi, which results in RBC fragmentation and hemolytic anemia. The marked increase in lactate dehydrogenase is attributed more to the massive tissue ischemia than to the RBC lysis. TTP may be acquired or familial. The acquired type is very heterogeneous, and the mechanisms that trigger the TTP pathophysiology are not completely clear. Approximately half of the patients present with secondary TTP associated with stem cell transplantation, pregnancy, disseminated cancer, autoimmune disorders, systemic infection, and use of certain drugs, including antiplatelet and chemotherapeutic agents.3,6,7 Of the remaining nonsecondary or idiopathic TTP cases, about 50% to 80% show a severe deficiency (less than 10% activity) of the VWF-cleaving protease known as a disintegrin-like and metalloprotease domain with thrombospondin type 1 motifs 13 (ADAMTS 13).6,7,9 Under normal conditions, ADAMTS 13 cleaves the long multimers of VWF into shorter segments as they are released from the endothelial cells, thus preventing excessive platelet adhesion, aggregation, and microthrombi formation.2,5 The ADAMTS 13 deficiency in acquired TTP has been attributed to inhibition by an immunoglobulin G (IgG) autoantibody, but inhibitory autoantibodies are not consistently found in all patients with the deficiency.6,10,11 For the remaining cases of idiopathic TTP, the underlying cause is not known.9 Approximately 80% of patients with idiopathic TTP respond favorably to plasma exchange therapy, likely due to the removal of the offending ADAMTS 13 autoantibody and infusion of replacement ADAMTS 13 enzyme from donor plasma.5,7 Therefore it is important that this type of TTP be recognized and quickly treated with plasma exchange therapy to avoid a fatal outcome.5 Approximately one third of patients who respond to plasma exchange experience recurrent episodes of TTP.3 Except when the TTP is related to autoimmune disease, pregnancy, or ticlopidine use, patients with secondary TTP generally do not respond well to plasma exchange, and the prognosis in these cases is poor.5,6,7,12 Familial TTPs are rare disorders due to mutations in the ADAMTS13 gene. Over 90 different mutations have been identified, and symptomatic individuals are either homozygous for one of the mutations or compound heterozygous for two different mutations.12 The mutations result in reduced ADAMTS 13 activity, usually 5% to 10% of normal.3 Familial TTP usually presents in infancy with recurrent episodes; however, some patients may not be symptomatic until adulthood after their system is stressed by pregnancy or a severe infection. Infusion of fresh frozen plasma supplies the deficient ADAMTS 13 enzyme.3 HUS is characterized by microangiopathic hemolytic anemia, thrombocytopenia, and acute renal failure from thrombi formed mainly in the glomerular microvasculature.5 There are two general types of HUS: D+ HUS, which is preceded by an episode of acute gastroenteritis, often with bloody diarrhea; and atypical HUS, which does not have antecedent diarrhea.5 Patients with HUS have the typical laboratory findings of microangiopathic hemolytic anemia discussed previously. In addition, there is usually a mild to moderate decrease in platelets and evidence of renal failure, including an elevated level of serum creatinine, proteinuria, hematuria, and the presence of hyaline, granular, and RBC casts in the urine (see Box 24-2). The D+ HUS type comprises 90% of cases of HUS.13 The most common cause is infection with enterohemorrhagic serotypes of Escherichia coli (such as O157:H7).3 D+ HUS occurs most often in young children, but can be found in patients of all ages. Patients initially have acute gastroenteritis, often with bloody diarrhea, and after 1 to 3 days develop oliguria and other symptoms of renal impairment. About one fourth of patients develop neurologic manifestations.13 E. coli serotypes implicated in HUS release Shiga toxins (Stx-1 and Stx-2) that are absorbed from the intestines into the plasma, and have an affinity for the Gb3 glycolipid receptors on endothelial cells, particularly those in the kidney, brain, and intestines.3,14 The toxin is transported into the endothelial cells, where it inhibits protein synthesis and causes major endothelial cell injury. Although the mechanisms are not known, the Shiga toxin, together with many cytokines secreted as a result of the infection, may also induce changes in endothelial cells that are prothrombotic, including expression of adhesion molecules and secretion of increased amounts of long VWF multimers.3,14 The end result is endothelial cell disruption, formation of platelet-fibrin thrombi that block the microvasculature in the glomeruli, and acute renal failure.3,14 Microthrombi can also form in other organs in addition to the kidney. There is no specific treatment for D+ HUS, but patients are provided supportive care as needed, including hydration, dialysis, and transfusions.13,14 The symptoms usually resolve spontaneously in 1 to 3 weeks, and the prognosis is favorable for most patients.13 Atypical HUS comprises about 10% of cases of HUS, is more commonly found in adults, and has a poor prognosis.12,15 Approximately half of the cases are familial and the other half are sporadic. The familial type is caused by mutations in genes that code for proteins involved in the alternative complement pathway or its regulation, such as the genes for factor H, factor I, factor B, membrane cofactor protein, and thrombomodulin.12,15 The mutations result in uncontrolled activation of the complement pathway, which causes endothelial injury, activation of platelets and coagulation factors, and formation of platelet-fibrin thrombi that obstruct the glomerular microvasculature.3,16 The sporadic form occurs secondary to some drugs, cancer, organ transplantation, pregnancy, and human immunodeficiency virus (HIV) infection.16 HELLP syndrome is a serious complication in pregnancy and is named for its characteristic presentation of hemolysis, elevated liver enzymes, and low platelet count. It occurs in fewer than 1% of all pregnancies, but develops in approximately 10% to 20% of patients with preeclampsia, most often in the third trimester.17 The exact pathogenesis is not known. In preeclampsia, abnormalities in the development of placental vasculature results in poor perfusion and hypoxia. As a result, antiangiogenic proteins are released from the placenta that block the action of placental growth factors, including vascular endothelial growth factor.3,18 The continued vascular insufficiency of the placenta results in maternal endothelial cell dysfunction, which leads to platelet activation and fibrin deposition in the microvasculature, particularly in the liver.18 Anemia, biochemical evidence of hemolysis, and schistocytes on the peripheral blood film are found as in the other thrombotic microangiopathies. The platelet count is less than 100 × 109/L; counts falling below 50 × 109/L indicate a worse prognosis.18,19 The level of lactate dehydrogenase is elevated, which reflects the hepatic necrosis as well as the hemolysis. The level of aspartate aminotransferase can be markedly elevated due to the severe hepatocyte injury. The low platelet count and increased levels of lactate dehydrogenase and aspartate aminotransferase are major diagnostic criteria for the HELLP syndrome and are used to assess the severity of the disease.17,18 The prothrombin time and the partial thromboplastin time are within the reference range, which distinguishes the HELLP syndrome from DIC. Therapy includes delivery of the fetus and placenta as soon as possible along with supportive care to control seizures, hypertension, and fluid balance. The mortality rate is 3% to 5% for the mother and 9% to 24% for the fetus.18 DIC is characterized by the widespread activation of the hemostatic system, resulting in fibrin thrombi formation throughout the microvasculature. The major clinical manifestations are organ damage due to obstruction of the microvasculature and bleeding due to the consumption of platelets and coagulation factors and secondary activation of fibrinolysis.20 DIC is a complication of many disorders, such as disseminated cancers, acute leukemias, infections, obstetric complications, crush or brain injuries, acute hemolytic transfusion reactions, extensive burns, snake or spider envenomation, and chronic inflammation (see Table 42-14). Thrombocytopenia of varying degree is a consistent finding. Only about half the patients have schistocytes on the peripheral blood film.4 The prothrombin time and partial thromboplastin time are prolonged, the fibrinogen level is decreased, and the level of D-dimer is increased in DIC, which distinguish it from the other thrombotic microangiopathies. Hypertensive crisis or hypertensive emergency (previously called malignant hypertension) is a severe and abrupt increase in blood pressure with acute organ damage, particularly in the heart, brain, and kidney.21 The sudden mechanical stress induced by the elevated blood pressure damages the endothelial cells and results in increased vascular permeability, activation of coagulation factors and platelets, and fibrin deposition in damaged vessel walls.21 The manifestations of hypertensive crisis include encephalopathy, acute myocardial infarction, aortic dissection, pulmonary edema, severe preeclampsia, acute renal failure, and microangiopathic hemolytic anemia.21,22 In one study, 27% of patients with hypertensive crisis had microangiopathic hemolytic anemia when the criteria were a platelet count of less than 150 × 109/L and either an elevated level of lactate dehydrogenase or the presence of schistocytes on the peripheral blood film.22 When the hypertension is brought under control, the RBC fragmentation disappears and the platelet count returns to normal within a few days.22,23 Mechanical hemolysis can occur in patients with prosthetic cardiac valves due to the turbulent blood flow through and around the implanted device.18,24 The hemolysis is usually mild, and anemia does not generally develop due to compensation by the bone marrow.24 Severe hemolysis is rare and is usually due to paravalvular leaks in prosthetic cardiac valves.24 Hemolysis can also occur in patients with cardiac valve disease prior to corrective surgery.18 The anemia that occurs in severe cases is usually normocytic, but can be microcytic if iron deficiency develops due to chronic urinary hemoglobin loss. Depending on the severity of the hemolysis and the ability of the bone marrow to compensate for the reduced RBC life span, patients can be asymptomatic or present with pallor, fatigue, and even heart failure.24 On the peripheral blood film, schistocytes are a characteristic feature due to the mechanical fragmentation of the RBCs (Figure 24-4). The reticulocyte count is increased, but the platelet count is within the reference range. Lactate dehydrogenase activity and levels of serum indirect bilirubin and plasma hemoglobin are elevated, whereas the serum haptoglobin level is decreased. Hemoglobinuria may be observed in severe hemolysis. Hemosiderinuria and a decreased level of serum ferritin occur with chronic hemoglobinuria due to the urinary loss of iron. Surgical repair or replacement of the prothesis may be required if the anemia is severe enough to require transfusions. For patients with hemoglobinuria, iron supplementation is provided to replace urinary iron loss. Folic acid may also be required, because deficiencies can occur due to the increased erythropoietic activity in the bone marrow.24 RBC hemolysis, with an increase in free plasma hemoglobin and a decrease in serum haptoglobin level, has been demonstrated after long-distance running and to a lesser extent after intensive cycling and swimming,25–27 but frank hemoglobinuria after exercise is a rare occurrence.28 Exercise-induced hemoglobinuria has been reported mainly in endurance runners, but has also been observed after strenuous hand drumming.25,29 Various causes have been proposed, including mechanical trauma from the forceful, repeated impact of the feet or hands on hard surfaces,25,26,29 increased RBC susceptibility to oxidative stress,30,31 and exercise-induced alterations in membrane cytoskeletal proteins.31,32 Exercise-induced hemoglobinuria does not usually cause anemia unless the hemoglobinuria is particularly severe and recurrent.28 Laboratory findings include a decreased level of serum haptoglobin, an elevated level of free plasma hemoglobin, and hemoglobinuria observed after strenuous exercise. Patients also have a slight increase in mean cell volume (MCV) and reticulocyte count, and with rare exceptions schistocytes are not observed on the peripheral blood film.28,29 Exercise-induced hemoglobinuria is a diagnosis of exclusion, and other possible causes of hemolysis and hemoglobinuria should be investigated and ruled out.28 There is no treatment for the disorder other than minimizing the physical impact on the feet with padding in shoes, running on softer terrain, or, if hemolysis is severe, discontinuing the activity. Malaria is a potentially fatal condition caused by infection of RBCs with protozoan parasites of the genus Plasmodium. Most human infections are caused by P. falciparum and P. vivax, but P. ovale, P. malariae, and a fifth species, P. knowlesi, also infect humans. P. knowlesi, a natural parasite of macaque monkeys, is easily misdiagnosed as P. malariae by microscopy, because the organisms are difficult to distinguish morphologically. Since 2004, hundreds of microscopically identified cases of P. malariae infection in Malaysia were actually found to be caused by P. knowlesi when polymerase chain reaction (PCR) assays were used, including tests on archival blood films from 1996.33–35 With the use of molecular techniques, P. knowlesi malaria is now known to be widespread in Malaysia, and cases have been reported in other areas of Southeast Asia.33–35 Half the world’s population (approximately 3.3 billion people) live in areas in which malaria is endemic and are at risk for the disease.36,37 Worldwide in 2008, there were an estimated 243 million cases of malaria with 863,000 deaths, mostly in children younger than 5 years of age.36 The majority of deaths were in Africa (85%), followed by Southeast Asia (10%) and the Eastern Mediterranean region (4%).36
Extrinsic Defects Leading to Increased Erythrocyte Destruction—Nonimmune Causes
Case Study
Patient Results
Reference Range
WBCs (×109/L)
11.0
4.5-11.0
Hb (g/dL)
8.7
12.0-15.0
Hct (%)
25
35-49
MCV (fL)
92
80-100
Platelets (×109/L)
176
150-450
Microangiopathic Hemolytic Anemia
Thrombotic Thrombocytopenic Purpura
Hemolytic Uremic Syndrome
HELLP Syndrome
Disseminated Intravascular Coagulation
Hypertensive Crisis
Macroangiopathic Hemolytic Anemia
Traumatic Cardiac Hemolytic Anemia
Exercise-Induced Hemoglobinuria
Hemolytic Anemia Caused by Infectious Agents
Malaria
Incidence
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Extrinsic Defects Leading to Increased Erythrocyte Destruction—Nonimmune Causes