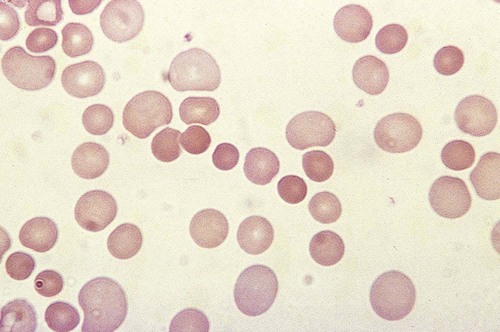
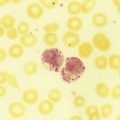
After completion of this chapter, the reader will be able to: 1. Define immune hemolytic anemia and indicate the types of antibodies involved. 2. Compare and contrast the mechanisms of immune hemolysis mediated by immunoglobulin M (IgM) and IgG antibodies. 3. Describe typical laboratory findings in immune hemolytic anemia and the importance of the direct antiglobulin test (DAT). 4. Compare and contrast four types of autoimmune hemolytic anemia in terms of the immunoglobulin class involved, the temperature for optimal reactivity of the autoantibody, the proteins detected by the DAT on the patient’s red blood cells, the presence of complement activation, the type and site of hemolysis, and the specificity of the autoantibody. 5. Relate results of the DAT to the pathophysiology and clinical findings in autoimmune hemolytic anemia. 6. Describe three mechanisms of drug-induced immune hemolysis. 7. Compare and contrast the pathophysiology of immune hemolysis due to drug-dependent and drug-independent antibodies, including the related laboratory findings. 8. Describe two types of hemolytic transfusion reactions, the usual immunoglobulin class involved, the typical site of hemolysis, and important laboratory findings. 9. Describe the cause, pathophysiology, and laboratory findings in Rh and ABO hemolytic disease of the fetus and newborn. 10. Given a patient history and results of a complete blood count, peripheral blood film examination, pertinent biochemical tests on serum and urine, and the direct and indirect antiglobulin tests, determine the type of immune hemolysis. The platelet count was within the reference range. Spherocytes and polychromasia were observed on the peripheral blood film (Figure 25-1). The film also revealed a slight increase in neutrophilic bands and 2 nucleated RBCs per 100 WBCs (not shown in figure). An increase in urobilinogen was noted on the routine urinalysis report, but the result for blood was negative. Urine and sputum culture results were negative. Significant findings from the chemistry profile included elevated levels of serum indirect bilirubin and lactate dehydrogenase. Subsequently, a direct antiglobulin test (DAT) and 3 units of red blood cells were ordered. A positive result on the DAT for IgG was reported. The indirect antiglobulin test on the patient’s serum and on an eluate prepared from the patient’s cells yielded negative results for all panel cells tested. 1. Is this type of anemia caused by an intrinsic or extrinsic defect? 2. What does the positive DAT result imply? 3. What mechanisms can lead to the development of drug-induced immune hemolytic anemia? 4. Describe the mechanism that is the most probable cause of this patient’s anemia. 5. In addition to the transfusion, what additional action should be taken? Immune hemolytic anemia and nonimmune hemolytic anemia are the two broad categories comprising the extrinsic hemolytic anemias, disorders in which red blood cells (RBCs) are structurally and functionally normal, but a condition outside of the RBCs causes premature hemolysis. The nonimmune extrinsic hemolytic anemias are the result of physical or mechanical injury to the RBCs and are covered in Chapter 24. The immune hemolytic anemias are conditions in which RBC survival is shortened due to an antibody-mediated mechanism. The antibody may be an autoantibody (directed against a self RBC antigen), an alloantibody (directed against an RBC antigen of another person), or an antibody directed against a drug taken by the patient (or its metabolite). Some antibodies are able to activate the classical complement pathway, which results in the attachment of activated complement proteins to the RBC membrane. RBCs with bound antibody or complement may be prematurely removed from the circulation extravascularly by macrophages (due to their receptors for complement and the Fc component of antibody), intravascularly by complement-mediated hemolysis, by or a combination of both mechanisms.1 Anemia develops when the amount of hemolysis exceeds the ability of the bone marrow to replace the RBCs lost. The degree of anemia varies from asymptomatic and mild to severe and life-threatening. The immune hemolytic anemias may be classified into the following groups: autoimmune hemolytic anemia, drug-induced immune hemolytic anemia, and alloimmune hemolytic anemia (Box 25-1).1,2 It is important to determine the cause of an immune hemolytic anemia so that the appropriate therapy can be administered to the patient. In immune hemolysis an antibody binds to an antigen on the surface of RBCs and that binding results in premature removal of those cells from the circulation through extravascular or intravascular hemolysis (see Chapter 22). The two classes or isotypes of antibodies involved in most immune hemolytic anemias are immunoglobulin G (IgG) and IgM. IgG is a monomer in a Y-like structure with two identical heavy chains (γ H chains) and two identical light chains (either κ or λ) connected by disulfide bonds.3 At the top of the Y-like structure are two antigen binding (Fab) domains, each formed from the N-terminus of the variable domain of one light and one heavy chain. IgG has one Fc domain (the stem of the Y) consisting of the C-terminus of the two heavy chains. IgM is a pentamer consisting of five monomeric units connected by disulfide linkages at the C-termini of their heavy chains (µ H chains).3,4 Because the composition, structure, and size of IgG and IgM are different, their properties and mechanisms in mediating hemolysis are also different. The classical complement pathway is an important mediator of immune hemolysis. The major proteins of the classical complement pathway are designated C1 through C9, and their components or fragments are designated with lowercase suffixes. The first protein, C1, has three components: C1q, C1r, C1s. After an antibody binds antigen on the RBC surface, C1q must bind to two adjacent Fc domains to activate the pathway.4 Theoretically only one IgM molecule is needed for activation due to its pentameric structure with five Fc domains; however, with the monomeric IgG, at least two molecules in close proximity are required.4 Therefore IgM antibodies are highly effective in activating complement, whereas IgG antibodies are unable to activate the pathway unless there is a sufficient number of IgG molecules on the RBC surface.4,5 In addition, subclasses IgG3 and IgG1 have high binding affinity for C1q, whereas subclasses IgG2 and IgG4 have minimal ability to bind complement.4,5 The binding of C1q to adjacent Fc domains activates C1r, which subsequently activates C1s. C1s activates C4 and then C2, which results in the binding of a small number of C4bC2a complexes to the RBC membrane. The C4bC2a complex is an active C3 convertase enzyme that cleaves C3 in plasma; this results in the binding of many C3b molecules on the RBC surface. C3b binds to C4bC2a to activate C5 to C5b. Next, C5b, C6, C7, C8, and C9 form the membrane attack complex (MAC). The MAC inserts into the lipid bilayer, forming a pore that allows water and small ions to enter the cell, which eventually causes intravascular lysis.3 Negative regulators inhibit various complement proteins and complexes in the pathway to prevent uncontrolled activation and excessive hemolysis.1,4 Hemolysis mediated by IgM antibodies requires complement and can result in both extravascular and intravascular hemolysis.5 When IgM molecules attach to the RBC surface in relatively low density, complement activation results in C3b binding to the membrane, but complement inhibitors prevent full activation of the pathway to the terminal membrane attack complex.1,5 C3b-sensitized RBCs are destroyed by extravascular hemolysis, predominantly by macrophages (Kupffer cells) in the liver, which have C3b receptors. Some of the C3b on the RBCs can be cleaved, however, which leaves the C3d fragment on the cell. RBCs sensitized with only C3d are not prematurely removed from circulation, because macrophages lack a C3d receptor.5 In severe cases of immune hemolysis involving heavy sensitization of RBCs with IgM antibody, significantly more complement is activated, which overwhelms the complement inhibitors. In these cases, complement activation proceeds from C1 to C9 and results in rapid intravascular hemolysis.5 Hemolysis mediated by IgG antibodies occurs with or without complement and predominantly by extravascular mechanisms.5 RBCs sensitized with IgG are removed from circulation by macrophages in the spleen, which have receptors for the Fc component of IgG1 and IgG3.1,5 IgG antibodies are not efficient in activating complement; therefore, intravascular hemolysis by full activation of complement from C1 to C9 is rare (except with anti-P in paroxysmal cold hemoglobinuria).5 However, if there is a high density of IgG3 or IgG1 bound to antigens on the RBCs, some complement is activated and C3b binds to the membrane. If both IgG and C3b are on the RBC membrane, there is faster clearance from the circulation by macrophages in both the spleen and liver.1,5 Often, IgG-sensitized RBCs are only partially phagocytized by macrophages, which results in the removal of some membrane. Spherocytes are the result of this process, and they are the characteristic cell of IgG-mediated hemolysis.5 The spherocytes are eventually removed from circulation by entrapment in the red pulp of the spleen (splenic cords), where they are rapidly phagocytized by macrophages (see Chapter 8).5 The mechanisms of immune hemolysis are summarized in Table 25-1. TABLE 25-1 Major Mechanisms of Immune Hemolysis Ig, Immunoglobulin; RBC, red blood cell. Laboratory findings in immune hemolytic anemia are similar to the findings in other hemolytic anemias and include decreased hemoglobin (Hb), increased reticulocyte count, increased levels of indirect serum bilirubin and lactate dehydrogenase, and decreased serum haptoglobin level. If the hemolysis is predominantly intravascular, the haptoglobin level will be moderately to severely decreased, plasma hemoglobin will be increased, and the patient may have hemoglobinuria or even hemosiderinuria (in cases of chronic hemolysis) (see Chapter 22). The mean cell volume may be increased due to the reticulocytosis, and leukocytosis and thrombocytosis may be present due to the increased erythroid proliferation in the bone marrow.1,6 Findings on the peripheral blood film include polychromasia (due to the reticulocytosis), spherocytes (due to IgG-mediated membrane damage by macrophages), and RBC agglutination (in some cases of IgM autoantibodies).5 Nucleated RBCs, occasional schistocytes, and erythrophagocytosis may also be observed on the peripheral blood film.1 To determine if the hemolysis is due to an immune mechanism, a direct antiglobulin test (DAT) is performed. The DAT detects in vivo sensitization of the RBC surface by IgG, C3d, or both.2 In the DAT procedure, polyspecific antihuman globulin (AHG) is added to saline-washed patient RBCs. Polyspecific AHG has specificity for the Fc portion of human IgG and complement component C3d and will agglutinate the RBCs if a critical number of one or both molecules is present on the RBC surface.2 If the DAT result is positive with polyspecific AHG, then the cells are tested with monospecific anti-IgG and anti-C3d to identify the type of sensitization. If IgG is detected on the RBCs, elution procedures are used to remove the antibody. The specificity of the antibody may be determined by assessing the reaction of the eluate with screening and panel reagent RBCs (RBCs genotyped for the major RBC antigens) using the indirect antiglobulin (AHG) test. Identification of any circulating alloantibodies or autoantibodies by the indirect antiglobulin test is also important in the investigation.2 The DAT result may be negative in patients with some immune hemolytic anemias.2 A positive DAT result has been reported in apparently healthy individuals and in blood donors with no evidence of hemolysis.7,8 In addition, other disorders beside immune hemolytic anemia can cause a positive DAT finding.2 Therefore diagnosis of immune hemolytic anemia cannot rely solely on the DAT and must take into account the patient history, symptoms, recent medications, previous transfusions, coexisting conditions including pregnancy, as well as the results of the applicable hematologic, biochemical, and serologic tests.2,5 Autoimmune hemolytic anemia (AIHA) is a rare disorder characterized by premature RBC destruction and anemia caused by autoantibodies that bind the RBC surface with or without complement activation. Autoantibodies may arise as a result of immune system dysregulation and loss of immune tolerance, exposure to an antigen similar to an autoantigen, B lymphocyte neoplasm, or other unknown reason.1,6 The type, amount, and duration of antigen exposure and genetic and environmental factors may also contribute to the development of autoantibodies.5,6 The anemia can be mild or severe, and the onset acute or gradual. The severity of the anemia depends on the autoantibody characteristics (titer, ability to react at 37° C, ability to activate complement, and specificity and affinity for the autoantigen), antigen characteristics (density on RBCs, immunogenicity), as well as patient factors (age; ability of the bone marrow to compensate for the hemolysis; function of macrophages, complement proteins and regulators; and underlying conditions).1,6 The autoimmune hemolytic anemias may be divided into four major categories based on the characteristics of the autoantibody and the mechanism of hemolysis: warm autoimmune hemolytic anemia, cold agglutinin disease, paroxysmal cold hemoglobinuria, and mixed-type autoimmune hemolytic anemia (Table 25-2).2,5,6 TABLE 25-2 Characteristics of Autoimmune Hemolytic Anemias Warm autoimmune hemolytic anemia (WAIHA) is the most commonly encountered autoimmune hemolytic anemia comprising up to 70% of cases.5 The autoantibodies causing WAIHA react optimally at 37° C, and the vast majority of them are IgG.1 WAIHA is most commonly found in adults older than 40 years of age and in children younger than 4 years of age.6 WAIHA may be classified as idiopathic or secondary. In patients with idiopathic WAIHA, the etiology is unknown. Secondary WAIHA may be found in many conditions such as lymphoproliferative diseases (chronic lymphocytic leukemia, B-lymphocyte lymphomas, Waldenström macroglobulinemia), nonlymphoid neoplasms (thymoma and cancers of the colon, kidney, lung, and ovary), autoimmune disorders (rheumatoid arthritis, scleroderma, polyarteritis nodosa, Sjögren syndrome, systemic lupus erythematosus), immunodeficiency disorders, and viral infections.1,6 The onset of WAIHA is usually insidious with symptoms of anemia (fatigue, dizziness, dyspnea), but some cases can be acute and life-threatening with fever, jaundice, splenomegaly, and hepatomegaly, especially in children with WAIHA secondary to viral infections.1,6 An underlying lymphoproliferative disorder is suggested in adults with massive splenomegaly, lymphadenopathy, fever, petechiae, ecchymosis, or renal failure.1,6 Although most autoantibodies that cause WAIHA are IgG, rare cases involving IgA autoantibodies as well as cases with fatal outcomes caused by warm-reacting IgM antibodies have been reported.1,2
Extrinsic Defects Leading to Increased Erythrocyte Destruction—Immune Causes
Case Study
Patient Results
Reference Range
WBCs (×109/L)
5.9
4.5-11.5
RBCs (×1012/L)
1.40
4.00-5.40
Hb (g/dL)
4.2
12.0-15.0
Hct (%)
13
35-49
Overview of Immune Hemolytic Anemias
Pathophysiology of Immune Hemolysis
IgM Mediated
IgG Mediated
Extravascular hemolysis
IgM activation of classical complement pathway from C1 to C3b only; clearance of C3b-sensitized RBCs by macrophages mainly in liver
Clearance of IgG-sensitized RBCs by macrophages mainly in spleen
Formation of spherocytes by partial phagocytosis of IgG-sensitized RBCs; spherocytes cleared by macrophages after entrapment in spleen
IgG* activation of classical complement pathway from C1 to C3b only; requires high-density IgG on RBC surface; clearance of IgG- and C3b-sensitized RBCs by macrophages in spleen and liver
Intravascular hemolysis
Full IgM activation of classical complement pathway from C1 to C9 and direct RBC lysis; requires high-density IgM on RBCs to overcome complement inhibitors
Full IgG activation of classical complement pathway from C1 to C9 and direct RBC lysis; requires very-high-density IgG on RBCs for activation and to overcome complement inhibitors; uncommon
Laboratory Findings in Immune Hemolytic Anemia
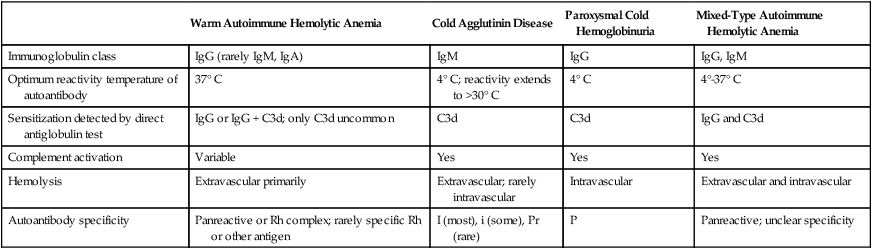
Autoimmune Hemolytic Anemia
Warm Autoimmune Hemolytic Anemia
Cold Agglutinin Disease
Paroxysmal Cold Hemoglobinuria
Mixed-Type Autoimmune Hemolytic Anemia
Immunoglobulin class
IgG (rarely IgM, IgA)
IgM
IgG
IgG, IgM
Optimum reactivity temperature of autoantibody
37° C
4° C; reactivity extends to >30° C
4° C
4°-37° C
Sensitization detected by direct antiglobulin test
IgG or IgG + C3d; only C3d uncommon
C3d
C3d
IgG and C3d
Complement activation
Variable
Yes
Yes
Yes
Hemolysis
Extravascular primarily
Extravascular; rarely intravascular
Intravascular
Extravascular and intravascular
Autoantibody specificity
Panreactive or Rh complex; rarely specific Rh or other antigen
I (most), i (some), Pr (rare)
P
Panreactive; unclear specificity
Warm Autoimmune Hemolytic Anemia
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Extrinsic Defects Leading to Increased Erythrocyte Destruction—Immune Causes