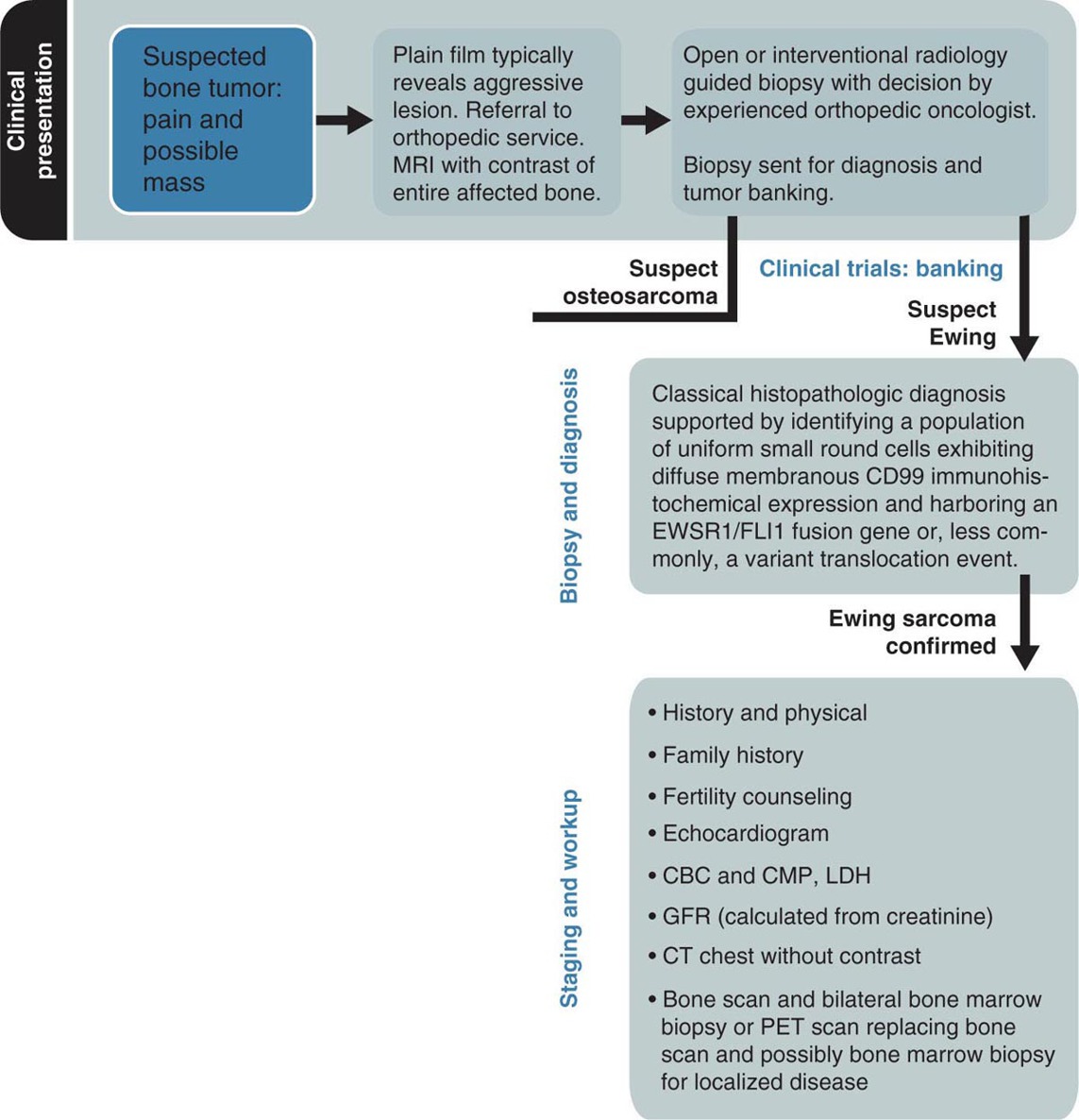
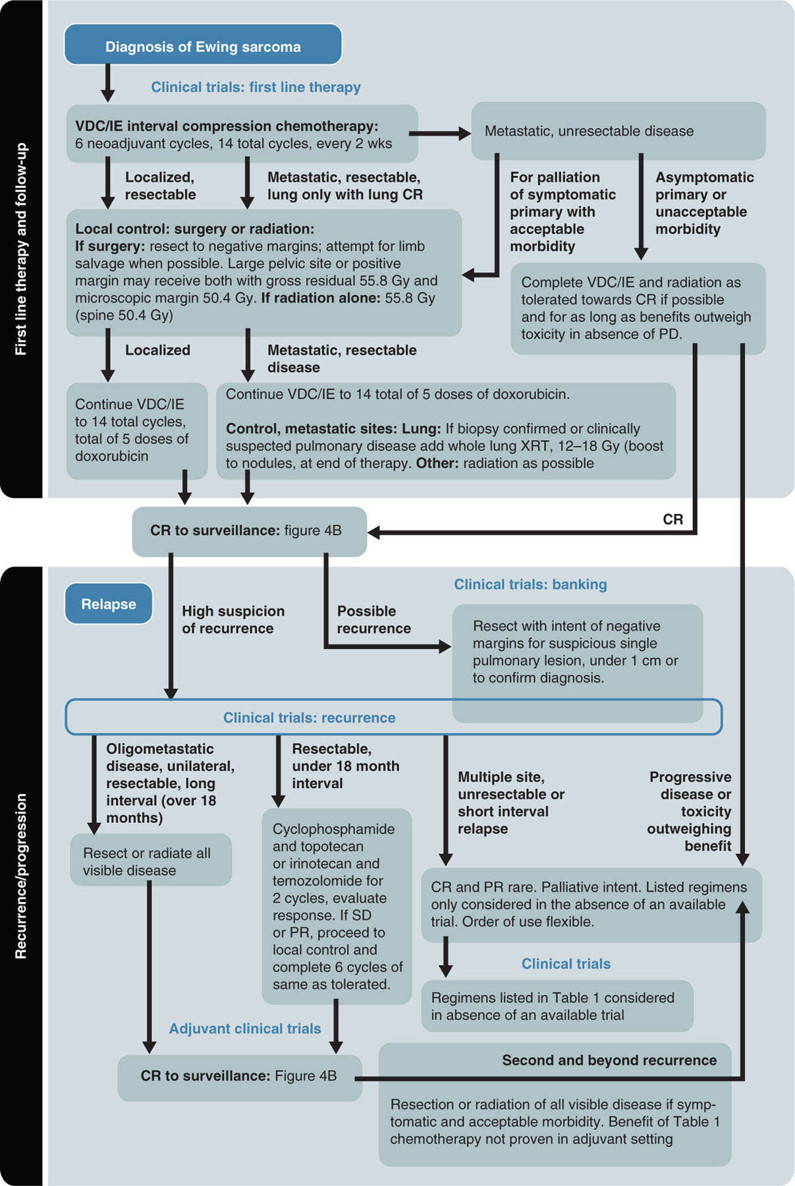
19914 Ewing Sarcoma The Ewing sarcoma (ES) family of tumors is a group of tumors consisting of typical ES, extraosseous ES, atypical ES, primitive neuroectodermal tumor, and Askin tumor. The unifying feature of these tumors is the specific chromosomal translocation fusing the EWS gene to a subset of ETS transcription factor family members, most commonly the FLI1 gene. The peak incidence of ES is during adolescence. Treatment of these tumors requires a multidisciplinary approach. The ideal team consists of experienced oncologists, orthopedists, radiation oncologists, radiologists, pathologists, and supportive care members. Optimal management of these patients by experienced teams is of particular importance. Given the lack of recent clinical progress, it is imperative to continue developing clinical trials and encouraging enrollment into these studies. This chapter discusses the current approach to diagnosis and management for those with the ES family of tumors. Ewing Sarcoma, Ewing Sarcoma family tumors, EWS-FLI1 translocation, Ewing Sarcoma treatment, Ewing Sarcoma chemotherapy Askin tumor, clinical trials, diagnosis, Ewing sarcoma, family members, management, primitive neuroectodermal tumor Clinical Trials as Topic, Diagnosis, Disease Management, Family, Neuroectodermal Tumors, Primitive, Sarcoma, Ewing INTRODUCTION The Ewing sarcoma (EWS) family of tumors is a group of tumors consisting of typical EWS, extraosseous EWS, atypical EWS, primitive neuroectodermal tumor (PNET), and Askin tumor. The unifying feature of these tumors is the specific chromosomal translocation fusing the EWS gene to a subset of ETS transcription factor family members, most commonly the FLI1 gene (less frequently, ERG, ETV1, E1A-F, or FEV). The peak incidence of EWS is during adolescence. Treatment of these tumors requires a multidisciplinary approach. The ideal team consists of experienced oncologists, orthopedists, radiation oncologists, radiologists, pathologists, and supportive care members. Despite extensive research leading to the finding of the disease-defining translocations, our understanding of the biology and its clinical correlation is still limited and there has been little progress in our ability to cure this disease in the past 30 years. Given the lack of recent clinical progress, optimal management of these patients by experienced teams is of particular importance. There have not been major advances in survival in patients with metastatic disease since standard use of adjuvant chemotherapy was adopted in the 1970s. Therefore, it is imperative to continue developing clinical trials and encouraging enrollment into these studies. This chapter will discuss the current approach to diagnosis and management for those with the EWS family of tumors. EPIDEMIOLOGY EWS is the second most common primary bone malignancy of childhood and young adulthood. About 80% of EWS occurs in patients younger than 20 years at the time of diagnosis.1 The annual incidence of EWS in this population is approximately 2.9 cases per million.2 The incidence is slightly higher in males than females at a ratio of 1.2:1. The most striking epidemiologic feature of EWS is the difference in racial distribution. It occurs more frequently in Caucasians, with a lower occurrence rate in people of Asian and African American (AA) descent.1,2 Worch et al. looked at the National Cancer Institute (NCI) Surveillance, Epidemiology, and End Results (SEER) database from 1973 to 2005 and found that the racial distribution of EWS was 78.6% white non-Hispanic, 14% Hispanic, 4.9% Asian, and 2.5% AA.3 This indicates lower occurrence in people of AA descent, as this group makes up about 13% of the population. This racial difference is seen at every age, and the explanation has yet to be fully elucidated. Beck et al., looking at the different length polymorphisms of the NR0B1 and CAV1 GGAA microsatellite in both European and African populations, were able to demonstrate that the number of GGAA microsatellite repeats is considerably higher in African populations, as a possible explanation.4 Additional research is needed to look into these racial differences and the correlation of the GGAA microsatellite repeats. CLINICAL PRESENTATION The presenting symptoms for EWS depend on the location of the tumor. When presenting in the bone, pain is a common presenting symptom, though a painless mass is not uncommon. When the initial site is in the soft tissue, the patient will often present with a mass, which may or may not be painful. Given that the primary incidence is in adolescence, pain is often mistaken for so-called “growing pains.” It is not uncommon for it to take weeks to months from the onset of symptoms for a diagnosis of EWS. EWS most often arises in the bones (85%), with the remaining 15% occurring in soft tissues.2 Additional presenting symptoms may include local swelling, pathologic fractures, and systemic symptoms such 200as fever and weight loss. EWS can be found in almost any bone or soft tissue. Approximately 38% of EWS appears at the diaphysis of the long bone in the extremities. EWS can present at the flat bones of the axial skeleton, such as the ribs and pelvis, at about 27%.2 Although extremity lesions are still more common, central lesions are seen in EWS more frequently. EWS that arises from the bone can also have a relatively large soft tissue component, especially when compared to osteosarcoma. There are no specific laboratory findings or tumor markers associated with EWS. Nonspecific findings such as an elevated serum lactate dehydrogenase (LDH) and alkaline phosphatase are seen with increased tumor burden, cell turnover, and bone destruction. Van Maldegem et al. completed a meta-analysis looking at possible biomarkers as prognostic factors.10 It was thought that LDH was a prognostic marker, but has been shadowed by the prognostic impact of metastatic disease, which clearly has the most impact on overall survival (OS). Decreases in white blood cells, hemoglobin, and platelets are not typically seen in EWS, unless there is significant bone marrow involvement. DIAGNOSTIC APPROACH Tissue diagnosis by an experienced pathologist is essential. Upfront resection of these tumors is not typically attempted in preference of biopsy, whether by percutaneous core-needle biopsy or via an open surgical procedure (Figure 14.1). The most common approach is to defer complete surgical resection until after the patient has received neoadjuvant chemotherapy. This has multiple benefits including allowing time for preparation for the surgery and obtaining custom prostheses if needed. Neoadjuvant chemotherapy typically also reduces the size of the tumor, which may facilitate surgical resection. Finally, neoadjuvant chemotherapy allows for immediate systemic therapy to treat/prevent the development of macroscopic and microscopic metastases. FIGURE 14.1 Workup for newly suspected bone sarcoma. CBC, complete blood count; CMP, complete metabolic panel; GFR, glomerular filtration rate; LDH, lactate dehydrogenase. Source: With permission from Reed DR, Hayashi M, Wagner L, et al. Treatment pathway of bone sarcoma in children, adolescents, and young adults. Cancer. 2017;123(12):2206–2218. doi: 10.1002/cncr.30589. 201Core-needle biopsy with image guidance can provide sufficient samples for diagnosis and, being less invasive, is the preferred initial biopsy technique.2 In rare cases, a core-needle biopsy does not provide enough tissue for diagnosis and a repeat or open biopsy must be attempted. It is imperative to minimize the tissue contamination from needle or open biopsy. The biopsy tract should be excised at the time of definitive surgery; hence, discussion with an experienced orthopedic oncologist on biopsy location is critical. If possible, the same surgical team performing the definitive surgery should obtain the biopsy or be consulted about the optimal image-guided core biopsy approach. Radiographic Features (Prior to Biopsy) At the time of diagnosis, a plain radiograph of the affected bone is done in addition to cross-sectional imaging, such as MRI or CT. In EWS, there will typically be x-ray findings in the diaphysis of the long bones that extend toward the metaphysis. The classic “onion skin” appearance of EWS is a lamellated periosteal reaction. It is seen in about 23% of presentations.1 More common nonspecific features seen on x-ray image include lytic lesions (75%), soft tissue extension (64%), and reactive bone formation (25%).1 Less common findings include radiating spicules, a.k.a. “sunburst appearance” (20%); periosteal thickening (19%); sclerosis (16%); and fracture (13%). The aforementioned sunburst appearance is more classically associated with osteosarcoma; however, it can be seen in both. The major difference between images of osteosarcoma and EWS is the location, which is typically at the metaphysis for osteosarcoma and diaphysis for EWS.1 Those with soft tissue EWS may have normal x-ray imaging, unless there is bony erosion or the soft tissue mass is large enough to be radiopaque. Once the initial x-ray study is completed, additional imaging is mandated as part of the workup. More detailed imaging of the primary site is needed using MRI with gadolinium contrast agent. This provides the resolution needed for proper surgical mapping prior to the biopsy and definitive surgery. In addition, a CT scan of the chest is required to assess for metastatic disease to the lungs, as it is the most common site for metastasis. Typically, a CT scan of the chest without intravenous (IV) contrast is sufficient, as the air provides the “contrast” needed when looking for metastatic tissue in the lungs; however, contrast may be needed if assessing the soft tissue component of an EWS involving the chest. Historically, technetium bone scan was used to assess for local and metastatic bone disease. The advantage to the bone scan is that it is cheaper and involves less radiation. However, more recently, there has been a shift to the fluorodeoxyglucose (FDG) positron emission tomography/computed tomography (PET/CT) as it is more sensitive for metastatic disease in EWS and can help assess nonskeletal lesions.2,5 MOLECULAR BIOLOGY The pathognomonic molecular finding in EWS family tumors is a reciprocal chromosomal translocation, most commonly occurring between the chromosomes 11 and 22. More specifically, t(11;22)(q24;q12) occurs in about 85% of cases. The gene name for the chromosome 22 translocation partner is EWSR1, and the gene name for the chromosome 11 partner is FLI1. The presence of this fusion protein correlates with a high expression of cell surface sialoglycoprotein CD99.2 Of the 15% of EWSs that do not harbor the EWSR1–FLI1 translocation, these tumors typically have evidence for gene rearrangements for EWSR1 to a different fusion partner, the most common being ERG on chromosome 21q22, which is seen in about 10% of EWS family tumors. Other less frequent fusion partners seen in less than 1% of cases include ETV1 on chromosome 7p22, ETV4 on chromosome 17q12, and FEV on chromosome 2q33. In addition to EWSR1-associated translocations, other chromosomal changes can also be simultaneously present. Trisomy 8 and 12 are observed in 50% and 33% of cases, respectively.2 Structural changes at chromosomes 1 and 16 are observed in about 20% of cases, typically leading to the gain of 1q, a loss of 16q, or the formation of a derivative chromosome der(1;16). There have been some reports describing fusions between FUS and either ERG or FEV in EWS. The FUS protein is highly related to EWS. The clinical significance of these various translocation partners to EWSR1, along with the other structural chromosomal changes, is unknown at this time. Additional research is needed to determine their significance. It is believed that a second mutational event is needed to immortalize these EWS–FLI1-expressing cells. It was initially thought that p53 pathway was involved, but aberrations in p53 are relatively rare 202in EWS, occurring in less than 15% of tumors. Some preclinical data suggest that NOTCH signaling or insulin-like growth factor 1 receptor (IGF1R) circuit is involved.2 There is no conclusive evidence that any of these varying aberrations, such as the varying translocation partners with EWSR1, such as FLI1, ERG, ETV1, E1A-F, or FEV gene, play a role in a clinically meaningful way in regard to prognosis. Currently, the main prognostic factor remains the presence or absence of metastatic disease. It is important to acknowledge that in Europe, they also look at the response to neoadjuvant chemotherapy, which at the time of writing this chapter had not yet been incorporated in North American protocols. THERAPEUTIC APPROACH EWS requires a multidisciplinary team approach to treatment, including orthopedic oncology, interventional radiology, radiation oncology, radiology, and medical/pediatric oncology (see Figure 14.2). A specialized orthopedic oncologist can take care of the biopsy and the surgical resection of the primary tumor. The biopsy could also be completed by interventional radiology with consultation of the primary surgical team to identify the proper approach to ensure removal of the biopsy tract when the patient undergoes definitive surgical resection of the primary tumor. Radiation oncology is involved to provide radiation for local control of the primary tumor site when needed if complete surgical resection is not possible, the patient is not a surgical candidate, the patient declines surgery, or there are positive margins after surgical resection. A pediatric or medical oncologist is involved to administer systemic chemotherapy. When available, patients should be encouraged to participate in clinical trials to further advance our treatment strategies and potentially receive improved treatment. Neoadjuvant chemotherapy should be initiated quickly after completing histologic confirmation of the diagnosis, metastatic workup, and baseline assessment. The use of chemotherapy to treat EWS started only in the 1960s. It was Sutow and Sullivan who, in 1962, published a report on cyclophosphamide to treat EWS.11 In 1974, Rosen et al. from Memorial Sloan Kettering first started treating EWS with multiple agents together, as is currently done.12 Their initial regimen consisted of vincristine, cyclophosphamide, actinomycin D, and doxorubicin (VACD). Chemotherapy greatly improved the outcomes of patients with EWS. The first cooperative study by Intergroup Ewing’s Sarcoma Study (IESS) from 1973 to 1978 showed that the addition of doxorubicin to VAC had a 5-year OS rate of about 60%, which was compared to 24% with VAC alone.6 The American Intergroup Ewing’s trial (INT-0091/POG-8850/CCG7881) determined that the addition of ifosfamide and etoposide (IE) to VACD improved the event-free survival (EFS) rate to 69% in those with localized disease.6 There was, however, no benefit in metastatic disease. The combination of cooperative groups in North America has allowed for increased enrollment of patients with this rare tumor into therapeutic clinical trials. The Children’s Oncology Group (COG) is one of the leading cooperative groups in pediatric oncology in North America. The COG trial AEWS0031 demonstrated improved survival in EWS patients with localized disease when treated with an interval-compressed version of VDC/IE given every 2 weeks instead of every 3 weeks. In Europe, a different standard regimen of VIDE is used, which consists of vincristine, ifosfamide, doxorubicin, and etoposide.5 The Euro-E.W.I.N.G. 99 clinical trial looked at the safety of treating with VIDE, compared to earlier standard of vincristine, actinomycin D, and cyclophosphamide (VAC) or vincristine, actinomycin D, and ifosfamide (VAI) in Europe. It demonstrated tolerance and efficacy, and thus, the Europeans now treat with VIDE (vincristine 1.5 mg/m2 × 1 day, ifosfamide 3 g/m2 × 3 days, doxorubicin 20 mg/m2 × 3 days, and etoposide 150 mg/m2 × 3 days). Currently, an ongoing Euro-Ewing 12 study (EudraCT number 2012-002107-17) is being conducted which looks at a direct comparison of VIDE and VDC/IE. Historically, the outcomes have been comparable between regimens; however, only now are they being compared head to head. The therapeutic regimen used in North America consists of vincristine (1.5 mg/m2, on day 1), doxorubicin (75 mg/m2 on days 1 and 2), cyclophosphamide (1,200 mg/m2 on day 1) alternating every 2 weeks with ifosfamide (1,800 mg/m2 on days 1–5) and etoposide (100 mg/m2 on days 1–5). This is referred to as VDC/IE and is considered the current standard of care for a pediatric oncologist in North America. It currently consists of 12 weeks of neoadjuvant chemotherapy, followed by local control with surgery and/or radiation and then an additional 16 weeks of adjuvant chemotherapy for a total of 28 weeks of therapy. While most pediatric oncologists follow the COG regimen described above, adult medical oncologists treat with different protocols. As demonstrated in the National Comprehensive Cancer Network (NCCN) Guidelines Version 1.2019, the possible regimens medical oncologists use to treat localized EWS are: VDC/IE (same as pediatrics), VDI (vincristine 2 mg/m2, doxorubicin 75 mg/m2, and 203ifosfamide 10 g/m2), and VIDE. When treating metastatic disease, they use the same three regimens plus the additional option of VDC alone. FIGURE 14.2 Pathway for treating newly diagnosed and relapsed Ewing sarcoma. CR, complete response; IE, ifosfamide and etoposide; PD, progressive disease; PR, partial response; SD, stable disease; VDC, vincristine, doxorubicin, and cyclophosphamide; XRT, x–ray therapy. Source: Used with permission from Reed DR, Hayashi M, Wagner L, et al. Treatment pathway of bone sarcoma in children, adolescents, and young adults. Cancer. 2017;123(12):2206–2218. doi:10.1002/cncr.30589.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


