Molecular Pathogenesis
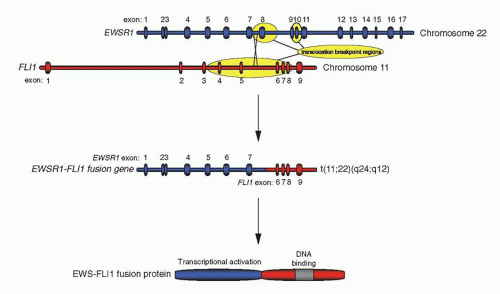
Gene rearrangements of
EWSR1-ETS and, rarely,
FUS-ETS are pathognomonic of Ewing sarcoma, and this suggests that these rearrangements play a fundamental mechanistic role in inducing malignant transformation associated with Ewing sarcoma. Indeed, expression of a full-length fusion product is a consistent finding in Ewing sarcoma. For example, although the majority of
EWSR1-FLI1 gene rearrangements result in the production of chimeric proteins with the EWS amino terminus fused to the carboxyl-terminal protein portion of FLI1, about one-third of gene rearrangements in Ewing sarcoma involve breakpoints in
EWSR1 exon 8 or intron 8, leading to out-of-frame fusions with
FLI1. In these cases, the reading frame is always restored, primarily by splicing out of exon 8, resulting in consistent expression of a functional full-length fusion protein in the tumors. This finding represents strong evidence for an essential role of EWS-ETS fusion proteins not only in the generation but also in the maintenance of the tumor. This assertion is also supported by two additional lines of experimental data: (a) EWS-FLI1 and related fusion proteins transform NIH3T3 (an immortalized murine fibroblast cell line) in vitro rendering it tumorigenic in nude mice
19 and (b) EWS-FLI1 antagonists (antisense RNA, antisense oligonucleotides, dominant negative constructs, small inhibitory RNA) block Ewing sarcoma cell growth in vitro and in human tumor xenografts in immunodeficient mice.
19,20,21The phenotype of tumors obtained in immunodeficient mice after transplantation of EWS-ETS-transformed NIH3T3 cells clearly differs from the phenotype after transformation with other EWS transcription factor fusions, resembling that of human Ewing sarcoma with partial neural differentiation. This suggests that the translocation product induces the Ewing sarcoma phenotype, rather than the phenotype resulting as a consequence of the cell-of-origin of the tumor. Such an assertion is also supported by the structure of fusion genes observed in human tumors with both EWSR1-CHOP and FUS-CHOP characterizing myxoid liposarcoma, and with EWSR1-TEC/CHN and TAF15-TEC/CHN found in extraskeletal myxoid chondrosarcoma. In this respect, FUS-ERG appears to be promiscuous, as it is present in both a small number of acute myeloid leukemias as well as a small number of Ewing sarcomas.
The minimal transforming protein portion involves at least the first 82 amino acids of EWS (encoded by the first four exons) and the DNA-binding domain of the ETS partner (in
FLI1 encoded by the last exon, exon 9). For full transformation potential, however,
EWSR1 exons 1 to 7 and, in
EWSR1-FLI1 fusions, inclusion of the very C-terminal domain of FLI1 are required.
19 In fact, despite significant variation in the architecture of chimeric EWS-ETS RNAs in Ewing sarcoma,
EWSR1 exons 1 to 7 and the DNA-binding domain encoding exons of the
ETS fusion partner are always present in the translocation products of Ewing sarcoma.
11 The most frequent gene fusions in Ewing sarcoma join
EWSR1 exon 7 to
FLI1 exon 6 or exon 5 in 51% and 27% of cases, respectively.
22The region encoded by the EWS portion of the fusion proteins is composed of 31 repeats of a degenerate hexapeptide motif, which, when combined with a DNA-binding domain, act as a potent transcriptional activation domain. Transcriptional activation may be related to the normal function of germline EWS that, like its close relatives FUS and TAF15, has been found associated with the RNA polymerase II complex and to interact with several RNA-processing proteins, possibly building a bridge between RNA transcription and maturation.
23 EWS- and FUS-transcription factor fusion proteins not only retain some of these interactions but also gain the ability to communicate with additional components of the RNA-processing machinery and to interfere with normal RNA splicing at least in in vitro assays.
23,24 Of particular recent interest is the demonstration that EWS and FUS can polymerize under some conditions to form fibers, and that these fibrous structures can interact with the carboxyl-terminal domain of RNA polymerase II.
25,26,27 This suggests that at some genetic loci, EWS-FLI1 may interact with RNA polymerase II through this polymerization mechanism to regulate gene expression. Interestingly, it has also been demonstrated that EWS-FLI1 binds to DNA sequences that contain repetitive GGAA-sequences (microsatellites), possibly creating the appropriate condition to allow these polymers to form and recruit RNA polymerase II.
27,28 Conversely, inhibition of at least some target genes is mediated by interactions between EWS-FLI1 and the Nucleosome Remodeling and Deacetylase corepressor complex, including its associated histone deacetylase and lysine-specific demethylase 1 proteins.
29 Thus, tumor-derived fusion proteins between EWS (or FUS) and the DNA-binding domain of a transcription factor are considered to exert much of their oncogenic activity via inappropriate regulation of target genes; some target genes are upregulated, while others are downregulated.
20,21 Expression of FET-ETS fusion genes leads to specific changes of the transcriptome and eventually to distinct cell type-dependent physiologic outcomes. For example, EWS-FLI1 induces cell death in primary mouse fibroblasts, which may either be rescued by loss of
p53 or
Ink4A gene function or a p53-dependent cell cycle arrest in immortalized human fibroblasts.
16,30 Pluripotent mesenchymal stromal cells are blocked
from further differentiation by ectopic EWS-FLI1 expression,
31,32 whereas transdifferentiation is observed in human neuroblastoma and rhabdomyosarcoma cell lines.
33,34 In contrast, established fibroblast cell lines are either transformed or remain unchanged in response to EWS-FLI1.
19,30 Model systems based on patient-derived Ewing sarcoma cell lines have been used to investigate EWS-FLI1 function in its native cellular context.
20,21,35 The gross changes in gene expression profiles induced by ectopic expression of EWS-ETS proteins in model systems and by EWS-ETS antagonists in Ewing sarcoma cells suggest that malignant conversion of the Ewing sarcoma precursor cell is the result of changes in the activity of a multitude of EWS-ETS downstream genes rather than of single genes. Some of the best-validated candidates include the upregulated genes
NKX2.2 and
NR0B1, and the repressed genes
IGFBP3 and
TGFBRII.20,21,35,36 In some cases, linkages to known molecular pathways important for oncogenesis are apparent (such as modulation of insulin-like growth factor and transforming growth factor signaling), but in other cases, novel oncogenic pathways appear to be involved.
The difference in fate of primary and immortalized cells ectopically expressing EWS-FLI1, cell death or survival with an altered or blocked differentiation program and transformation suggests the presence of a second mutational event in Ewing sarcoma. Although in experimental systems abrogation of the p53 pathway by either
p53 mutation or
INK4A deletion cooperates with EWS-FLI1 in transformation by stabilizing EWS-FLI1 expression,
30 these aberrations are relatively rare in Ewing sarcoma, occurring with a frequency of less than 15% for each.
14 The p53-dependent DNA damage response pathway has been shown to be largely intact in Ewing sarcoma.
37,38 The p53 protein also plays a role as a guardian against oncogenic stress, and induction of cell death by ectopic EWS-FLI1 expression in primary fibroblasts may reflect this function. It, therefore, remains to be demonstrated why most Ewing sarcomas tolerate EWS-ETS oncogene expression in the presence of wild-type p53. Inhibition of NOTCH signaling may be involved
39 or alternately autocrine growth factor loops may be involved, such as the insulin-like growth factor receptor 1 (IGF1R) circuit. Transformation of rodent fibroblasts by EWS-FLI1 has been shown to require IGF1R expression
40; EWS-FLI1 inhibits expression of IGFBP3,
20 which increases IGF1R signaling; and inhibition of IGF1R interferes with Ewing sarcoma growth in nude mice.
41,42,43 The possible roles of other autocrine and paracrine growth factor pathways that have been found to be active in Ewing sarcoma, including platelet-derived growth factor beta, gastrin-releasing peptide, neuropeptide Y and stem cell factor, remain unknown.
44,45,46,47
Histogenesis
The histogenesis of Ewing sarcoma has been a matter of controversy ever since the first description of Ewing sarcoma as a diffuse endothelioma of bone by James Ewing in 1921,
48 and has subsequently been ascribed to endothelial, mesenchymal, and hematopoietic stem cells based on ultrastructural features.
49 Further immunocytochemical, molecular studies, and in vitro differentiation of Ewing sarcoma cell lines using agents such as retinoic acid and dibutyryl adenosine cyclic monophosphate provided evidence for the neural differentiation potential of Ewing sarcoma. This was initially interpreted as an argument for a descent from neural crest tissue. Comparative gene expression profiling studies support this view and reveal a unique signature that clearly distinguishes Ewing sarcoma from other small round cell tumors.
50 Because of the ability to synthesize choline acetyltransferase and the lack of appreciable synthesis of adrenergic pathway precursors, Ewing sarcoma could be derived from postganglionic parasympathetic primordial cells located throughout the parasympathetic autonomic nervous system. However, the observation that EWS-FLI1 expression in murine fibroblasts imposes a partial neural phenotype on the transformed cells and that introduction of the fusion protein into human neuroblastoma cells switches the adrenergic phenotype to the cholinergic pattern of Ewing sarcoma suggest a role for the chromosomal translocation in determining the pattern of differentiation marker expression in this disease.
33 EWS-ETS fusion proteins are transcription factors that probably bind to the same sequence motifs in the human genome as those recognized by their normal ETS counterparts. It is therefore possible that Ewing sarcoma at least partially recapitulates the phenotype of tissues that express either FLI1 or ERG during normal development, such as hematopoietic tissues, endothelial cells, and neural crest cells.
51,52An alternative hypothesis regarding the histogenesis of Ewing sarcoma is that it might arise from mesenchymal stem cells.
53 Mesenchymal stem cells can be found in the bone marrow-derived stromal cell compartment and have the ability to differentiate into multiple distinct lineages, including bone, cartilage, and fat cells. Initial studies demonstrated that expression of EWS-FLI1 in either murine bone marrow-derived stromal cells, or in myoblasts, blocked those cells’ ability to differentiate in tissue culture.
31,32 Subsequent studies found that inhibition of EWS-FLI1 expression in patient-derived Ewing sarcoma cell lines caused those cells to adopt a mesenchymal stem cell phenotype.
53 This phenotype included a gene expression pattern that was similar to normal mesenchymal stem cells, the expression of characteristic mesenchymal stem cell surface markers, and the ability to differentiate into adipogenic, chondrogenic, and osteogenic lineages. Attempts to model Ewing sarcoma through the introduction of EWS-FLI1 into human fibroblasts or mesenchymal stem cells have recapitulated the gene expression pattern of bona fide Ewing sarcoma but have not resulted in oncogenic transformation, suggesting that additional events are required for this process.
16,54 Ultimately, the final proof of the long sought-after Ewing sarcoma cell of origin may have to await the development of animal models to test these hypotheses explicitly.
Biologic Determinants of Prognosis
At diagnosis, patients with Ewing sarcoma either present with localized disease or clinically overt metastases. In the prechemotherapy era when local measures (surgery or radiation) were the only modes of therapy, more than 90% of patients died from tumor dissemination regardless of their stage at presentation.
55 With current multimodal treatment regimens, however, more than two-thirds of patients with localized disease can be cured. Survival rates of patients with metastatic disease remain very poor despite modern multimodal therapy. In addition to stage (localized vs. metastatic), a series of other putative prognostic factors have been investigated.
The availability of a unique and specific tumor marker, the
EWSR1-ETS gene rearrangement, allows for high-sensitivity tumor cell detection in tissues and body fluids by molecular techniques. Retrospective studies suggest that RT-PCR positivity of bone marrow for
EWSR1-ETS rearrangement may be of prognostic relevance not only in patients with overt metastases but also in those with localized disease.
56,57 However, this finding has not yet been confirmed prospectively. Previous studies suggested that the EWS-ETS RNA fusion type (exon composition) was prognostic.
22,58 However, two prospective studies failed to demonstrate significant differences in clinical outcomes for patients with
EWSR1-FLI1 type 1,
EWSR1-FLI1 type 2, or
EWSR1-ERG-translocated tumors.
59,60Among cytogenetic aberrations frequently accompanying the tumor-specific translocation of chromosome 22, controversial results have been obtained in terms of the potential prognostic impact of the gain of chromosomes 8 and 12.
61,62 Gain of 1q and loss of 16q have recently been described as associated with adverse outcome.
61,62,63 Patients with tumors that have more complex karyotypes or with hyperdiploidy exceeding 50 chromosomes may have a worse outcome.
64,65 This may also be true for loss of 1p, which
is associated with poor prognosis in neuroblastoma, in which it occurs at a much higher rate than in Ewing sarcoma.
61 Among the aberrations commonly associated with many cancers, aneuploidy,
TP53 mutation (encoding p53 protein),
INK4A gene deletion (encoding the p16 and ARF proteins), and telomerase expression have been described as unfavorable prognostic markers in Ewing sarcoma, whereas P-glycoprotein expression has been found to be of no significance for the course of the disease.
66,67,68,69,70 All of these factors were evaluated in small, retrospective case series and have not been confirmed prospectively.
A range of other markers have been proposed to be prognostic, but have not yet been validated. For example, at least two studies have evaluated gene expression platforms to identify prognostic signatures.
71,72 CD56 coexpression was reported to be an adverse prognostic factor in one study.
73 Another group reported that higher baseline serum IGF-1 levels were associated with a more favorable outcome,
74 though a prospective study failed to validate this finding.
75Additional clinical variables have also been evaluated for possible prognostic impact. Histologic evaluation of response to neoadjuvant chemotherapy is a prognostic factor in Ewing sarcoma, although applicable only to patients who undergo surgical resection after neoadjuvant chemotherapy.
76,77 Response to chemotherapy can also be assessed by magnetic resonance imaging (MRI).
78 Fluorine-18 fluorodeoxyglucose positron emission tomography (FDG-PET) may provide a useful tool for monitoring response to therapy and outcome.
79
Targeted Therapy
The ideal tumor-specific target would be the EWS-ETS fusion protein or critical gene products regulated by the fusion protein. Experimentally, proof of principle has been obtained by both antisense and RNA interference studies that demonstrated that modulation of EWS-FLI1 expression results in growth inhibition of Ewing sarcoma in vitro and in vivo.
79,80,81,82,83 The clinical use of antisense RNA oligonucleotides or small inhibitory RNAs is impeded by the difficulty of efficiently delivering nucleic acids into disseminated tumor cells. One possible method to achieve this goal may be the inclusion of oligonucleotides into nanocapsules or nanospheres. This approach has successfully halted Ewing sarcoma growth in xenotransplanted nude mice.
82,84 Another approach has been to identify available drugs, which may directly or indirectly modulate the expression of EWS-FLI1. One group performed a high-throughput screen and demonstrated that cytarabine downregulates the expression of EWS-FLI1.
85 This preclinical finding did not result in meaningful clinical activity in a phase II study of cytarabine in patients with refractory Ewing sarcoma.
86 Interfering with the interaction of the EWS-ETS fusion protein with RNA helicase A is another potential targeted strategy, with a derivative compound from broad screening, YK-4-279, showing xenograft inhibition.
87CD99 may represent another promising candidate for targeted therapy in Ewing sarcoma. Although neither a ligand for CD99 nor the mechanisms by which this antigen is involved in Ewing sarcoma are known, in vitro studies on cell lines demonstrated that CD99 binding and silencing by specific antibodies induces rapid tumor cell death, enhanced by combination with conventional chemotherapeutic drugs. In vivo studies, which to date have been restricted to athymic mice subcutaneously xenografted with a Ewing sarcoma cell line, have shown inhibition of Ewing sarcoma growth following treatment with anti-CD99.
88 However, there is no direct homolog of CD99 in mice, and thus, toxicity of anti-CD99 treatment cannot be assessed in this model. Because of high-level expression of CD99 in human hematopoietic stem cells, endothelial cells, and several cell types in the gonads and the pancreas, clinical trials using anti-CD99 antibodies remain untested.
Angiogenesis, the formation of new blood vessels, is crucial to the progression of malignant disease, because otherwise the tumor would rapidly outgrow its nutrient supply.
89 A striking feature of Ewing sarcoma is the presence of large blood lakes that are lined by tumor cells, potentially due to tumor cell plasticity under hypoxic conditions.
90 The EWS-ETS oncoprotein activates the vascular endothelial growth factor (VEGF) promoter, both directly and through an interaction with the transcription factor Sp1.
91 Seventeen of 31 Ewing sarcoma tissue samples were immunostain positive for VEGF, and had an inferior outcome when compared with the 14 whose tumors were negative (
p = 0.0047).
91 Similarly, serum VEGF was elevated in a small series of patients with Ewing sarcoma at the time of initial diagnosis.
92 The endogenous antiangiogenic peptide, thrombospondin, is downregulated in Ewing sarcoma.
93 However, low-dose antiangiogenic chemotherapy, with vinblastine and celecoxib, when combined to standard chemotherapy, did not improve outcome in 35 patients with metastatic Ewing sarcoma compared to historic results.
94Several lines of evidence support that targeting IGF1R will be an effective strategy in Ewing sarcoma. Almost all Ewing sarcoma cell lines have expression of both IGF1 and IGF1R.
95,96 Neutralizing antibodies against the IGF1R inhibit the growth of multiple Ewing sarcoma cell lines.
41,95,96 These findings have also been observed in Ewing sarcoma mouse xenografts.
41 Clinical trials of IGF1R antibodies have been disappointing, with 6% to 14% response rates, although some responses have been durable.
97,98,99,100 In vitro studies have demonstrated that treatment with an IGF1R monoclonal antibody markedly increases the sensitivity of Ewing sarcoma cells to chemotherapy, supporting future combination studies.
101