Abstract
The appropriate measurement of hormones, their interpretation, the judicious use of provocative tests, and targeted imaging are all important aspects of the discipline of reproductive endocrinology. In this chapter only clinically relevant tests have been included. Some algorithms have been provided suggesting a differential diagnosis for the evaluation of some disorders, but only to illustrate how testing plays an important role in making a correct diagnosis. Details of pathophysiology and treatment for specific disorders may be found in other chapters in this text.
Keywords
Assays, LH, FSH, GnRH, prolactin, estrogens, progesterone, androgens, AMH, inhibins, ovarian reserve, HCG, insulin resistance, adipokines, GH, bone markers, vitamin D, thyroid hormones, CRH, ACTH, adrenal, CT, MRI, ultrasound
Introduction
This chapter reviews the assessment of hormonal status in the practice of reproductive endocrinology. It is acknowledged that the patient will often provide useful biologic information, such as changes induced by hypoestrogenism, skin changes with androgen excess, and galactorrhea in hyperprolactinemia. The clinically relevant symptoms and signs associated with various disorders are discussed in other chapters in this text. Here we will describe various types of hormonal assays and dynamic tests used in the evaluation of reproductive disorders, as well as diagnostic radiographic techniques. We also offer several algorithms for the diagnosis of common clinical disorders with the use of hormonal assessments where necessary.
Principles of Hormone Assays
- ◆
An understanding of the principles of all immunoassays is necessary to understand their usefulness and limitations.
- ◆
There are several types of immunoassays as well as detection systems, some of which have been developed for rapidity and automation.
- ◆
The gold standard for the measurement of steroid hormones, which may be at very low circulating levels, is with the use of mass spectrometry (MS).
Immunoassays
The predominant assays used to measure steroid, peptide, and protein hormones in serum, plasma, and urine samples for over 45 years have been immunoassays. These assays have been widely used in both clinical diagnostic and research settings.
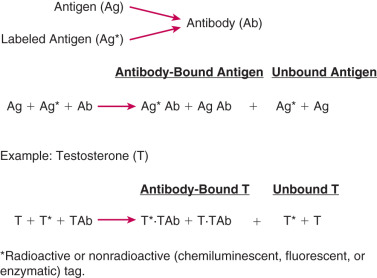
As the term immunoassay implies, this method of measuring hormones involves an antigen-antibody reaction, where the antigen is the hormone to be measured and the antibody, which is prepared against the hormone, binds to the hormone. The hormone can be a steroid, protein, or peptide. There are two types of hormone immunoassay methods: one uses excess hormone and a limited amount of antibody, whereas the other uses excess antibody. For quantification purposes, immunoassay methods require a labeled marker, which can be the radioactive [usually iodine-125( 125 I)] or nonradioactive (chemiluminescent, fluorescent, or enzymatic) form of the hormone being measured, or a suitable chemical derivative of the hormone.
General Principles of Immunoassays
Principle of Antigen-Excess Hormone Immunoassays
The principle of an antigen-excess type of hormone immunoassay involves competition between the hormone being measured and the labeled form of the hormone for a limited amount of the antibody prepared against the hormone. When all three components are combined in a test tube, the net result is a mixture of labeled and unlabeled hormone bound to the antibody, and unbound labeled and unlabeled hormone ( Fig. 34.1 ). From the practical standpoint in the assay, the bound hormone is separated from unbound hormone and the activity of the bound fraction is quantified (e.g., by determining the counts per minute of radioactive iodine in a gamma counter if a 125 I-labeled hormone is used as the marker). Separation of hormone-bound and unbound antibody is achieved by one of a variety of different methods, including use of a second antibody (prepared against the first antibody) when an iodinated hormone is used as the labeled antigen, or by magnetic particles when a nonradioactive hormone is used. In this manner, by using different concentrations of a pure hormone (standard), one can first generate a standard curve of the hormone. As the concentration of the standard is increased, the antibody-bound labeled marker is displaced; the higher the amount of standard that is added, the lower the amount of marker will be obtained. Thus, in an antigen-excess immunoassay, the standard curve shows an inverse relationship between the different amounts of antibody-bound labeled antigen (hormone) and the different concentrations of the standard ( Fig. 34.2 ). A standard curve is essential in any immunoassay method used to measure the levels of a hormone in a biological fluid.
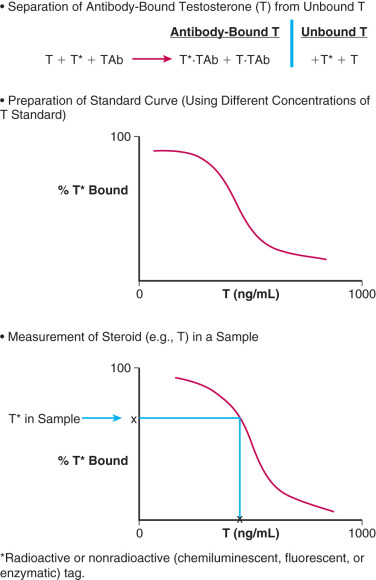
Measurement of a hormone in serum, plasma, or urine by an antigen-excess immunoassay is obtained by first determining the amount of the hormone that is antibody-bound in the sample. The concentration of the hormone is then extrapolated off the standard curve, as shown in Fig. 34.2 . An appropriate computer program can be used to generate the data rapidly.
Principle of Antibody-Excess Hormone Immunoassays
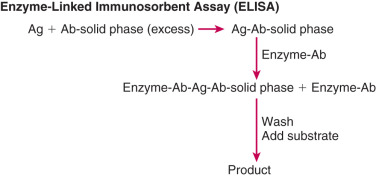
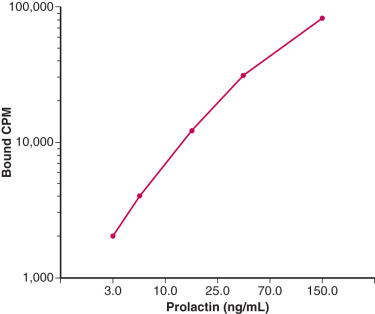
In contrast to antigen-excess immunoassays, which use a limited amount of antibody resulting in competition between labeled and unlabeled antigens, there is no competition between these antigens in antibody-excess immunoassays (often referred to as immunometric assays [IMAs]). In general, two different antibodies that recognize two different parts of the antigen are used in antibody excess immunoassays, forming a “sandwich,” with the antigen in the middle. Thus this assay method is useful for large molecules such as protein and peptide hormones. Quantitation is achieved by labeling one of the antibodies with a radioactive or nonradioactive marker. One of the most widely used antibody-excess immunoassays is the enzyme-linked immunosorbent assay (ELISA) in which quantitation is achieved by attaching an enzyme to one of the antibodies and measuring the optical density of a product formed by reaction of the enzyme with a specific substrate ( Fig. 34.3 ). When one of the antibodies is labeled with a radioactive tracer, this type of assay is referred to as an immunoradiometric assay (IRMA), and when the labeling involves a chemiluminescent or fluorescent tag, it is referred to as an immunochemiluminometric assay (ICMA) or immunofluorometric assay (IFMA), respectively. In contrast to the inverse relationship between antibody-bound labeled antigen and concentration of antigen in the standard curve in an antigen-excess immunoassay, there is a direct relationship between the labeled antibody and antigen concentration in an antibody-excess immunoassay ( Fig. 34.4 ).
Major Components of Immunoassay Systems
There are three major components of immunoassay methods: the antibody, unlabeled antigen (standard or substance to be measured in a biological fluid sample), and labeled antigen. These are essential in every immunoassay and are discussed below.
Antibody
The antibody is perhaps the most important component of an immunoassay method. If an antibody in an immunoassay recognizes only the compound that it is intended to measure, then it is likely that the assay will be highly accurate.
Antibodies are commonly produced in animals (polyclonal) or via cell culture (monoclonal). The typical protocol for production of polyclonal antibodies is to disperse a small amount of antigen into an adjuvant (e.g., Freund adjuvant) and to inject it intradermally at multiple sites into an animal such as the rabbit. After approximately 3 months, a blood (serum) sample is obtained from the animal and the antibody titer is determined. This procedure involves addition of a fixed amount of labeled antigen to serially diluted aliquots of the serum obtained from the animal. The antibody dilutions typically start at 1 : 1000 and cover a range of at least two orders of magnitude. After the bound antigen is separated from the unbound antigen, the dilution at which the antibody binds between 30% and 70% (usually 40% to 50%) of the total activity of the labeled antigen is used for the assay.
To obtain a monoclonal antibody, it is first essential to inject the antigen into a mouse to induce an immunological reaction in the spleen. Each immunized spleen cell can produce an antibody with specific characteristics. The most important step in the production of monoclonal antibodies is screening the spleen cells to separate those capable of secreting a single antibody type. When a myeloma cell from the same species of mouse is fused with the selected spleen cell, a hybridoma is produced. This hybridoma continuously secretes antibodies with the same characteristics as the selected spleen cell. A hybridoma cell is capable of producing hundreds of specific antibody molecules per second. Thus a continuous clone line can be maintained in culture, becoming a source for the production of homogeneous monoclonal antibody molecules.
Antigen
Here the term antigen can refer to the substance that is injected into an animal to produce an antibody or to the standard that is used to generate a standard curve in an immunoassay. In addition, antigen can refer to the substance that is being measured in a biological fluid (e.g., serum).
Antigen Used to Prepare an Antibody
In general, any molecule larger than 10,000 Da can elicit an antibody response. With molecules in the 1000- to 10,000-Da range, eliciting an immune response becomes more difficult. Molecules smaller than 1000 Da are generally nonantigenic and are coupled to a large protein molecule (e.g., albumin); these small molecules are referred to as haptens. Thus steroids and some peptides, but not proteins, are prepared as hapten-proteins and injected into the animal to produce a polyclonal or monoclonal antibody.
The site of attachment of the protein carrier (e.g., albumin) is important because it determines the specificity of the antibody. In general, highly specific antibodies are formed when the attachment site does not involve any functional group on the steroid molecule. A commonly used approach is to prepare a carboxyl derivative at the carbon-6 position of the steroid molecule and to couple it to an amino group of the protein carrier.
Antigen Used as Standard
Steroids and peptides are available commercially and in most instances can now be obtained in a relatively pure state. However, it should never be assumed that these products are 100% pure when developing an immunoassay. If necessary, the compounds can be purified (e.g., by use of high-performance liquid chromatography [HPLC]). In contrast, proteins are generally considered to be more difficult to purify.
Labeled Antigen
Use of an optimum labeled antigen is also important in an immunoassay, as this determines how the compound being measured in a biological fluid, such as serum, is quantified in the assay. The labeled antigen must not only be identifiable by some physical or chemical method (e.g., a radioactivity counter or spectrophotometer) but also must bind to the antibody.
The classical labeled antigens for immunoassays have been radioactive antigens. For example, insulin, the first hormone measured for a radioimmunoassay (RIA), was labeled with radioactive iodine ( 131 I). This isotope had a very short half-life (8 days) and was soon replaced with iodine-125 ( 125 I), which has a half-life of 60 days. Peptides have also been labeled with 125 I. For steroids, tritium ( 3 H) was the initial isotope used in RIAs, but was subsequently replaced with 125 I. It is important to realize that, unlike proteins and peptides, the steroid molecule itself cannot be iodinated. Instead, the steroid molecule is attached chemically to an iodinated carrier molecule such as histamine or tyrosine methyl ester.
To eliminate the high cost of storage and disposal of radioactive material and to allow immunoassays to be carried out on an automated platform, nonradioactive labeled antigens began to be used in these assays. The predominant labels that were attached to antigens were chemiluminescent, fluorescent, and enzyme (which required a substrate); these labels were detected by use of a luminometer, fluorometer, and spectrophotometer, respectively. Presently, there is wide use of the chemiluminescent label in immunoassays on automated platforms and the enzyme label in ELISAs carried out manually.
Automated Immunoassays
Over the past 30 years or so, highly sophisticated automated immunoassay systems have been developed. These systems consist of instruments that not only detect spectral properties of nonradioactive ligands (e.g., chemiluminescence), but also are able to analyze multiple analytes as well as to process multiple samples rapidly and efficiently. These immunoassay systems are capable of using both antigen-excess and antibody-excess methodology.
Currently a widely used immunoassay system is the Immulite analyzer (Siemens Healthcare Diagnostics, Deerfield, Illinois). The Immulite system employs enzyme-amplified chemiluminescent technology. The mechanism involves hydrolysis of a stable chemiluminescent substrate through the action of the enzyme alkaline phosphatase, resulting in an unstable anion that gives rise to sustained emission of light. The emitted light is quantified by use of a luminometer in the instrument.
As an example, the Immulite luteinizing hormone (LH) assay uses a solid-phase, two-site ICMA. The solid phase consists of a polystyrene bead coated with a monoclonal antibody against LH. The bead is sealed into an Immulite test unit, to which LH standard or serum sample, together with a polyclonal antibody conjugated to alkaline phosphatase, are added simultaneously. During an incubation period, LH is bound to the monoclonal antibody coating the bead and the polyclonal antibody-enzyme conjugate, forming a “sandwich complex.” After unbound conjugated antibody is removed by a wash, the amount of complex, which is directly proportional to the LH standard or LH in the sample, is quantified by use of the chemiluminescent substrate described previously.
Steroid Hormone Immunoassays
The first RIA was developed in 1969 by Guy Abraham and that was for measurement of estradiol (E 2 ) in serum or plasma. The E 2 RIA method involved separation of E 2 from interfering metabolites by organic solvent extraction and Celite or Sephadex column chromatography, prior to its quantitation by RIA. The purification steps were added to remove most of the estrogen metabolites. E 2 is readily converted to estrone (E 1 ) and both E 2 and E 1 are converted to a total of approximately 90 metabolites. The extraction step removes the conjugated (water-soluble) steroids, whereas the chromatographic step separates E 2 from potential interfering unconjugated metabolites. The E 2 RIA included a specific antiserum against RIA in conjunction with tritiated E 2 , and separation of the antibody-bound and unbound E 2 fractions using charcoal. This method, which is often referred to as a conventional RIA, was shown to be sensitive, specific, precise, and accurate. Soon afterward it was applied successfully to other sex steroid hormones, such as testosterone and progesterone.
During the decade of the 1970s, RIAs were developed for a variety of natural and synthetic steroid hormones. A notable change in the RIA method during those years was replacement of tritium with iodine in the labeled antigen to improve assay sensitivity. Because of their relatively low cost, RIAs became widely used in diagnostic and research laboratories.
Impact of the Conventional Steroid RIA Method
The immediate impact of the conventional steroid RIA method was that it allowed measurement of an immensely wide range of compounds of clinical and biological importance, and it opened new horizons in endocrinology. The long-term impact of the steroid RIA method was that its use in numerous studies enriched the field of endocrinology with new knowledge, and its use in diagnostic testing provided physicians with valuable information for diagnosing and treating countless patients. The steroid RIA methodology also allowed substantial research into the physiological and pathophysiological roles of steroid hormones in applications such as sexual differentiation, puberty, neuroendocrinology, the menstrual cycle, pregnancy, menopause, and male endocrinology. In addition, the RIA method opened the door for epidemiologic studies that permitted us to better understand the role of steroid hormones in the etiology of numerous diseases, notably the hormone-dependent breast and prostate cancers.
Advantages and Disadvantages of Conventional Steroid Radioimmunoassays
Steroid RIA methods with purification steps have the following advantages: First, steroid-binding proteins (BPs) (e.g., sex hormone-binding globulin [SHBG]) are denatured, thereby releasing the steroids (e.g., E 2 and testosterone) that they bind; second, the purification steps remove numerous potentially interfering metabolites prior to RIA; third, the RIAs are accurate and reliable when properly validated; finally, multiple steroids (usually up to five) can be measured in a single aliquot of serum.
Conventional steroid RIAs also have disadvantages. They are cumbersome, time-consuming, costly, and require relatively large sample volumes, especially when the steroid is present in low concentrations. Also, although multiple steroids can be measured in a single aliquot of serum, the measurements have to be done very carefully and are especially time-consuming. In addition, as with all antibody-based assays, since the measurement of the analyte is a surrogate approach (i.e., radioactivity is measured rather than the actual analyte itself), there is always the possibility of antibody cross-reactivity giving an erroneous result. Furthermore, the presence of autoantibodies within patients can further affect an assay, leading to falsely high or low values depending on the type of antibody interaction that occurs. Despite these concerns, a well-validated RIA preceded by organic solvent extraction and chromatography is typically an accurate and precise assay for the majority of applications.
Direct Steroid Immunoassays and Their Advantages and Disadvantages
Due to the time-consuming limitations of conventional RIAs, in the late 1970s the radio ligands in RIAs were replaced with nonradioactive ligands (chemiluminescent, fluorescent, or enzymatic) and the organic solvent extraction and chromatography steps used prior to RIA were eliminated, allowing direct immunoassays to be performed on an automated platform; this resulted in rapid measurements of steroid hormones. Conventional RIAs continued to be used, but their use was overwhelmingly surpassed by direct immunoassays, particularly in clinical diagnostic laboratories.
The development of automated platforms gave direct assays, such as chemiluminescent immunoassays, the advantages of being convenient, simple, rapid, and relatively inexpensive, and requiring a lower sample volume (usually 0.1 mL). However, these assays also have serious disadvantages. They often overestimate the measurements due to lack of specificity of the antibody, especially in samples obtained from women treated with exogenous steroid hormones. Also, matrix differences may exist between serum samples (particularly hemolyzed and lipemic samples) and solutions of the standard used to prepare the standard curve in the assay. In addition, steroids such as testosterone and E 2 may not be released efficiently from proteins such as SHBG to which they bind with high affinity in blood. Furthermore, direct immunoassays generally lack the sensitivity to measure accurately low levels of certain steroid hormones such as E 2 and testosterone. Limitations of direct immunoassays for quantifying E 2 in postmenopausal women and testosterone in both premenopausal and postmenopausal women are now well documented in the literature.
Protein and Peptide Hormone Immunoassays
The first protein hormone RIA was developed for insulin and preceded the first steroid hormone RIA, which was for E 2 , by about 10 years. The basis for the first protein RIA method was described in 1959 by Yalow and Berson, who showed that 131 I-insulin could be displaced by nonradioactive insulin from insulin-binding protein, and that 131 I-insulin was inversely and quantitatively related to the total amount of insulin present. This is the same principle that was described earlier for the steroid hormone RIA, and was subsequently used to develop RIAs for protein hormones other than insulin, as well as for peptide hormones. In 1967, Odell and coworkers reported the first RIAs for LH and follicle stimulating hormone (FSH). Subsequently, RIAs were developed for a variety of other protein hormones, such as human chorionic gonadotropin (hCG), prolactin and thyroid-stimulating hormone, and for peptide hormones such as gonadotropin-releasing hormone (GnRH) and adrenocorticotropic hormone (ACTH). The protein and peptide hormone RIAs used specific antisera in conjunction with the relevant 125 I-proteins and peptides. The change from the 131 I label to the 125 I label provided a longer-life for the labeled material (8 days vs. 60 days, respectively), so that the proteins and peptides did not require iodination as often for the RIAs.
Immunoradiometric Assay
In the late 1960s and early 1970s, the IRMA method was developed for measurement of proteins, which included insulin, FSH, and LH. Subsequently, the IRMA was also used to measure peptides such as GnRH and ACTH. The IRMA differed from the RIA in that the compound to be measured was assayed directly in combination with a specific labeled antibody rather than in competition with a labeled antigen for a limited amount of antibody.
Current two-site IMAs depend on the use of two antibodies directed to distinct antigenic epitopes on the protein or peptide being measured. This assay method is ideal for use of monoclonal antibodies, thereby increasing assay specificity. In addition, two-site IMAs offer greater sensitivity because they are much less dependent on the affinity of the antibody than RIAs. An appropriate choice of antibodies directed to different antigenic sites can further enhance the affinity and thereby the sensitivity of the assay. Thus highly sensitive IMAs opened up new clinical diagnostic opportunities (e.g., use of the sensitive thyroid stimulating hormone (TSH) assay to diagnose hypothyroidism).
Two-site IMAs also have certain limitations. They cannot be used to measure small ligand molecules such as steroids and very small peptides since they display only one antigenic site at a time. Also, IMAs are liable to high-dose “hook” effects. This results from very high levels of antigen that saturate the binding sites on the capture antibody and still leave antigen free to bind to the tracer antibody.
Enzyme-Linked Immunosorbent Assay
The ELISA differs from the IRMA in that the ELISA uses a solid-phase procedure and an enzyme-labeled instead of a radioactively labeled antibody. The most common format used for the ELISA is the multiwell plate made of polystyrene or polyvinyl. This is convenient to handle since centrifugation is not required and multiple wash steps are readily automated. Antibody is coated on the wall of each well and this antibody binds the corresponding antigen in the test sample. The antigen-antibody complex is quantified by addition of an enzyme-labeled specific antibody. Following addition of the appropriate substrate, the endpoint (product) can be “read” spectrophotometrically, using an automated reader.
Standards Used in Protein Immunoassays
Unlike steroids and small peptides (e.g., GnRH), proteins are considerably more difficult to purify. Although some proteins such as insulin are available in a highly purified state for use as standards in immunoassays, others such as FSH, LH, and hCG have not yet been prepared in sufficient amounts in a highly purified form. Although the latter proteins are available from international agencies, their purity, based on biological potency per unit weight, is often less than that of highly purified preparations reported by individual investigators. To be able to compare results obtained in different laboratories and at different times in the same laboratory, considerable effort has been made to use a single material as a standard.
Reference materials are available for FSH, LH, and hCG. They are provided by the World Health Organization (WHO), which obtains them from the National Institute for Biological Standards and Controls in Hertfordshire, England. Two types of reference materials are available: namely, an International Standard (IS) and an International Reference Preparation (IRP). The IS is a material that has a potency established in 10 to 20 expert laboratories throughout the world, whereas the potency of an IRP is established by only several laboratories.
With the advent of RIAs for gonadotropins, a partially purified extract of human pituitary glands (code name LER-907) was made available by the National Institutes of Health in the United States. This preparation was provided in large amounts to the WHO, which purified it further in 1976 and called it the First International Reference Preparation of Pituitary FSH and LH (1st IRP-FSH and LH). Subsequently, another partially purified extract of LER-907 was prepared in 1980 to replace the 1st IRP-FSH and LH. It was called the Second International Reference Preparation of Pituitary FSH and LH (2nd IRP-FSH and LH) and was assigned the code number 78/549. Highly purified preparations of pituitary FSH and LH have also been prepared. The First International Reference Preparation for Human Pituitary LH (1st IRP-LH, code number 68/40) was available in 1974. Subsequently (in 1988), this material was replaced by the Second International Standard for Pituitary LH (2nd IS-LH; code number 80/552). In 1986, the First International Standard for Pituitary FSH (1st IS-FSH, code number 83/575) was established. Presently, both the 2nd IRP-FSH and LH and 2nd IS-LH are used as standards in gonadotropin immunoassays.
The initial standard used in hCG immunoassays was a partially purified hCG preparation obtained from urine of women in their first trimester of pregnancy and was called the Second IS for hCG. Subsequently, a highly purified preparation was developed and was referred to as the Third IS. This preparation is presently used as the standard in hCG immunoassays.
Validation of Immunoassays
Before an immunoassay can be used to measure any compound, it must first be validated with respect to sensitivity, specificity, accuracy, and precision. Procedures used to validate immunoassays are described later.
Sensitivity
The sensitivity of an assay is defined as the smallest amount of the substance being measured that can be distinguished from zero. In practice, assay sensitivity depends on the precision of the standard curve, which can be measured by assaying 10 replicates of each concentration of standard and the “zero” standard, which contains no standard. This allows calculation of the mean ± standard deviation (SD) amount of the antibody-bound marker, corresponding to each concentration of standard. The sensitivity of an assay is the lowest standard concentration that yields a mean amount (e.g., counts per minute) of antibody-bound marker differing by two SDs from the mean amount of antibody-bound marker associated with the zero standard.
Assay sensitivity expressed in that manner, in practical terms, refers to the sensitivity of the standard curve. However, it is important to know the assay sensitivity in terms of the lowest amount of analyte that can be measured per milliliter of sample. This can be determined by calculating the product of the lowest standard concentration (based on standard curve sensitivity) and, if applicable, dilution factors, as well as the factor accounting for procedural loss (in assays using an extraction and/or chromatographic step).
The sensitivity of an assay is especially important when very low serum levels of a compound are being analyzed (e.g., measurement E 2 in samples from postmenopausal women or from patients treated with an aromatase inhibitor).
Specificity
Assay specificity refers to the degree of interference from cross-reaction that is encountered from substances other than the target analyte. The specificity of an immunoassay is usually assessed in two different ways. First, the cross-reactivity (expressed as a percentage) of the antibody is determined by comparing the dose-response standard curve of the substance being measured with dose-response curves obtained for compounds that may be present in the same sample as the analyte, and that may bind to the antibody. The percent cross-reaction of an antiserum with a substance is calculated from the mass of standard that yields 50% inhibition of binding of the assay marker to the antibody, divided by the mass of the cross-reacting substance that gives the same percentage of inhibition (50%), and multiplied by 100%.
A second approach for defining immunoassay specificity is to compare the analyte values of a group of samples measured by an immunoassay method with those obtained by a classic method such as MS. If this is not possible and the analytes are being measured in an immunoassay without chromatography, the analyte values should be compared with the values obtained by an immunoassay that uses an extraction step followed by a chromatographic step (e.g., Celite column partition chromatography) to separate the analyte in question from interfering metabolites.
Accuracy
Assay accuracy defines the extent to which a given measurement agrees with the actual value. One commonly used method to establish the accuracy of an immunoassay is based on the finding of linearity (parallelism) between the assay standard curve and serial dilutions (using assay buffer) of several samples containing known high concentrations of the analyte. Another method is based on the recovery of added (“spiked”) standard, at different levels, from patient samples. This method is often used for assays that require one or more purification steps (e.g., conventional steroid hormone assays). Both the parallelism and the “spiked” sample methods are limited by the precision of carrying out the dilutions or “spiking.”
Precision
The precision of an assay refers to the variability that exists when multiple measurements of the compound are made on the same sample. In practice, both intraassay precision and interassay precision are determined and are usually expressed as the coefficient of variation of replicate measures. The coefficient of variation (expressed as a percentage) is calculated by dividing the SD by the mean of replicate determinations of an analyte, and then multiplying by 100. Intraassay precision is assessed by measuring the analyte in replicate samples (usually 7 to 10) within the same assay. Interassay precision is determined by measuring the analyte in replicate samples (at least five), with each sample included in a different assay. Both intraassay and interassay precision should be determined in three different concentrations of the analyte (high, midrange, and low).
Interferences in Immunoassays
Circulating human antibodies that react with animal proteins (anti-animal antibodies) are often an unrecognized and unsuspected source of interference in immunoassays. This is particularly true with two-site immunoassays. The most common human anti-animal antibody interference is caused by human antimouse antibodies (HAMA). HAMA can cause either positive or negative interference in two-site mouse monoclonal antibody-based immunoassays. Strategies for preventing the development of anti-animal antibodies have been proposed. Awareness of laboratory personnel and clinicians of the problems associated with this type of interference in immunoassay methods is important.
Mass Spectrometry Assays
Measurements of steroid hormones by MS assays actually preceded their measurements by RIA. As early as 1966, the first comprehensive urinary steroid profile was produced by gas chromatography-mass spectrometry (GC-MS). This method combines the resolving power of gas chromatography with the high sensitivity and specificity of the mass spectrometer. Separation of steroids by gas chromatography requires that they be first derivatized to increase their volatility, selectivity, and detectability. The mass spectrometer functions as a unique detector that provides structural information on individual solutes as they elute from the gas chromatography column. The MS technique first involves ionization of the compound being measured at the ionization source, and is followed by separation and detection of the ions in the mass analyzer. A mass spectrum is produced in which the relative abundance of a particular ion is plotted as a function of the mass-to-charge (m/z) ratio, and the concentration of the compound is then obtained.
Due to the complexity of the methodology and associated costs of the instrumentation and reagents, as well as the need for a highly trained individual to carry out the assays, use of GC-MS assays was restricted to a limited number of laboratories, primarily at pharmaceutical companies. Thus, for a period of about 30 years after the development of the first RIA in 1969, conventional RIAs and direct immunoassays were the predominant methods used to quantify steroid hormones in clinical diagnostic and research laboratories due to their relative ease of performance and considerably lower overall costs. However, advances in liquid chromatography (LC) technology in the 1980s led to the development of a high performance liquid chromatography-MS (LC-MS) instrument in 1987. In addition, invention of an electrospray source by Nobel laureate, John B. Fenn, in 1990, and the subsequent development of chemical ionization greatly improved routine analysis of steroids. This technology facilitates ionization of compounds present in liquid droplets and sprays the molecules directly into the mass spectrometer from the HPLC unit. The advancements allowed for simple coupling of the LC eluent with the mass spectrometer and often negated the need for derivatizing the steroid, thereby reducing the complexity of the assay and shortening the assay run time dramatically. These factors greatly increased the throughput of patient samples, while still providing highly accurate and precise results.
In recent years, there has been a large increase in the use of assays that use either LC or gas chromatography with tandem MS (LC-MS/MS or GC-MS/MS). Tandem MS consists of two mass spectrometers in series connected by a chamber (collision cell). After chromatography, the sample is processed in the first mass spectrometer to obtain the precursor ion, which is then fragmented in the collision cell into product ions. The mass of the product ions is then determined in the detector of the second mass spectrometer. This method has high specificity, sensitivity, and throughput.
During the past decade there has been a substantial increase in the use of LC-MS/MS assays, particularly in major diagnostic clinical laboratories, and to a lesser extent in some research laboratories. However, there are still situations where a GC-MS assay provides higher chromatographic resolution and even sensitivity than LC-MS. A particular strength of GC-MS and GC-MS/MS assays is their high applicability to measurement of large numbers of structurally similar compounds. They remain the most powerful assay method for defining defects in steroid hormone metabolism.
Because of the high validity and throughput of steroid hormone MS assays, there is a rapidly growing use of this methodology in both clinical and research laboratories. In larger reference laboratories, these assays have replaced the conventional RIAs, which are cumbersome and time-consuming, and direct immunoassays, which lack specificity and/or sensitivity. The MS technology has been implemented successfully for routine analysis of steroid hormones in major clinical diagnostic laboratories. Although the high cost of MS instrumentation, related operating costs, and requirement for high technical expertise have prohibited smaller laboratories from using this instrumentation for high-throughput routine testing of steroid hormones, this situation is changing and MS assays are becoming much more widely used.
In addition to use of the MS methodology for routine analysis of steroid hormones, this methodology is now sufficiently rapid and robust for measuring these hormones in large epidemiological studies with high specificity and sensitivity. An important advantage of MS assays is their capability of measuring multiple steroids in a single aliquot of serum or urine. In contrast, generally only up to five different steroids can be measured in a single serum aliquot (usually 1 mL) by conventional RIA. Several years ago, at the National Cancer Institute, LC-MS/MS assays were developed for quantifying as many as 15 different estrogens in only 0.5 mL of serum or urine. In addition LC-MS/MS assays have been established for simultaneous quantitation of 11 androgens, including principal adrenal and gonadal androgenic precursors and their 5α-reduced metabolites.
Studies using MS have shown that the adrenal steroid, 11β-hydoxyandrostenedione, is a precursor to 11-ketotestosterone and 11-keto-5α-dihydrotestosterone, which are potent androgenic androgens. With LC-MS/MS several 11-oxygenated androgens (11β-hyroxyandrostenedione, 11-ketoandrostenedione, 11β-hydroxytestosterone, and 11-ketotestosterone) have been found to be elevated in women with polycystic ovary syndrome (PCOS) and cumulatively constitute a greater proportion of total circulating androgens than DHEA, androstenedione, and testosterone. More work on the clinical significance of these MS-based 11-oxygenated androgens is clearly needed.
Measurement of metabolite profiles of a steroid hormone has the potential to provide highly valuable information in diagnosing patients and in a variety of studies, particularly epidemiological studies. Quantitative analysis of metabolite profiles of small-molecular-weight molecules such as steroid hormones is referred to as metabolomics and is a rapidly growing field of research.
Standardization of Steroid Hormone Assays
Although hormone assays are widely used by physicians, some of the assays lack accuracy. Significantly different results can be obtained by two laboratories measuring the same hormone in the same serum sample from a patient. Inaccurate test results can lead to missed diagnosis or incorrect treatment. In the research setting, inaccurate tests can make findings in a study uncertain and not repeatable. This makes it very difficult to apply meaningful evidence for clinical decisions and establishing normal ranges. Accurate hormone assays will lead to fewer medical errors, eliminating the need for costly test repeats, and reducing overall costs of health care.
Although there seems to be general agreement that MS assays will become the “gold standard” for steroid hormone measurements, there are many challenges to overcome before this occurs. It is important to realize that the MS technology faces variability issues similar to those of conventional RIAs and direct immunoassays that need to be addressed. Differences in accuracy among MS assay methods appear to be attributable to variation in calibration of assay standards, whereas differences in assay precision may be explained, at least in part, by variations in sample preparation.
The Centers for Disease Control and Prevention (CDC), with broad input from various professional societies, particularly the Endocrine Society, is leading a Hormone Standardization Program with an initial focus on standardizing testosterone measurements. The conceptual approach of this program is built on experiences gained from successful standardization programs that the CDC maintains or has supported (e.g., the cholesterol standardization program and national glycohemoglobin standardization program). The testosterone standardization program consists of three basic steps: calibrating individual assays, developing a reference system, and verifying end-user test performance. Although it is evident from previous efforts at the CDC that vast improvements in measurement performance can be achieved through assay standardization, based on their experience it could take years to accomplish such achievements.
Reference Intervals of Steroid, Protein, and Peptide Hormones
An important requirement in clinical diagnostic testing in which measurements of steroid, peptide, or protein hormones are determined by conventional and direct immunoassays or MS assays is reference intervals. These intervals should be derived from well-characterized, adequate-sized populations using standardized procedures such as those formulated by the Clinical and Laboratory Standards Institution. Frequently, only limited information is available about subjects used to establish reference intervals. Also, an often neglected aspect in establishing reference intervals for hormone measurements is biologically influencing factors that may affect these measurements. The major factors include gender, age, body mass index (BMI), pubertal stage, menopausal status, phase of menstrual cycle, pregnancy, and diurnal rhythm.
It is especially important to compare reference intervals for steroid hormones measured by MS assays to those obtained by conventional RIAs, since most of our knowledge about the role of steroid hormones in normal women and men, as well as in various endocrine diseases, is based on the data obtained by the latter methodology. Presently it appears that the reference intervals obtained by the two methods do not differ significantly.
Measurement of Specific Reproductive Protein and Peptide Hormones and Steroid Hormones
- ◆
Sensitive assays are now available for the measurement of gonadotropins that have in large part alleviated the need to depend on stimulation tests, which are still required on occasion; the greatest need is with amenorrhea and puberty alterations.
- ◆
Prolactin may circulate in several molecular forms including macromolecules. An understanding of this is important in the evaluation of prolactin secreting adenomas.
- ◆
Estradiol is the most important estrogen measured for clinical purposes and may require MS for the required sensitivity at low levels.
- ◆
Progesterone is primarily used for the detection of ovulation.
- ◆
Androgen measurements are used for evaluation of androgen excess in women and hypogonadism in men. Several derivatives of routine androgens are now detectable using MS. The work up of androgen excess is outlined.
Measurement of Gonadotropins
Serum concentrations of LH and FSH are expressed in international units, using as a reference partially purified pituitary hormone preparations such as LER-907 or urinary gonadotropins (Second International Reference Preparation—human menopausal gonadotropin [2nd IRP-HMG]). The use of different hormone reference preparations complicates comparisons between different assays.
Classic RIAs of gonadotropins have low specificity and sensitivity and often cannot distinguish low levels from low-normal values. This is also compounded by the pulsatile nature of gonadotropin secretion. Immunometric assays are more sensitive and have improved the measurement of gonadotropins. As previously described, IRMA (immunoradiometric), IFMA, or ICMA methods should be used when low levels of gonadotropins are expected but may not offer advantages when elevated or normal levels are anticipated. The ELISA for gonadotropins is considered to be less sensitive and exhibits higher nonspecific binding.
Additional problems that may arise in LH measurements include a significant cross-reactivity with hCG and the pulsatile secretion pattern (pulses approximately every 60 to 90 minutes). The requirement of precision in values would necessitate that samples, taken 15 to 20 minutes apart, be pooled for analysis. In the clinical setting, however, this is not necessary because low levels will usually remain low, and high values will not usually be in the normal range. The concern about pulsatility is more for research purposes and is far less pronounced for FSH samples, where pulses are less frequent, occurring approximately every 3 hours.
Apart from alterations in FSH and LH associated with various disorders and physiologic states, FSH levels are increased by levodopa and ketoconazole and decreased by the administration of estrogens and phenothiazines. LH is increased by ketoconazole and decreased by administration of sex steroids, phenothiazines, digoxin, and propranolol.
Blood Levels of Luteinizing Hormone and Follicle-Stimulating Hormone in Women
In adult women, using conventional RIA assays, during the follicular phase, serum levels of LH and FSH by RIA range between 4 and 15 mIU/mL (2nd IRP-HMG standard). Third generation commercial assays, using immunometric methods, report lower normal ranges of LH (1.0 to 8 mIU/mL).
During mid-cycle, serum LH levels increase four- to sixfold while serum FSH levels increase two- to threefold. The mid-cycle increase of gonadotropins lasts about 2 days.
During the luteal phase, serum LH and FSH levels are similar to or slightly lower than their respective levels during the follicular phase. Therefore, in healthy women the LH:FSH ratio is about 1.0 during the follicular phase but increases during ovulation.
Before puberty, gonadotropin levels are lower than in adult women, and the ratio of LH:FSH is less than 1.0 because of the relative predominance of FSH secretion.
During the perimenopausal period, serum FSH levels increase substantially while LH levels remain in the normal range. After menopause, both gonadotropins are high but serum FSH levels are higher than LH. In older women, LH levels decrease from their high levels in early menopause. In general a ratio of LH:FSH less than 1.0 reflects a hypoestrogenic state.
Gonadotropin Stimulation Tests (Gonadotropin-Releasing Hormone and the Gonadotropin Releasing Hormone Agonist)
In the past, synthetic GnRH, the hypothalamic decapeptide, has been widely used to stimulate LH and FSH secretion to uncover abnormalities that cannot be diagnosed with baseline determinations. However, in most instances, the test largely reflects baseline measurements.
In patients with PCOS and women with ovarian failure, GnRH stimulation results in exaggerated increases in LH (in the setting of PCOS) and in FSH elevations in women with ovarian failure. The GnRH test may be useful in the differential diagnosis of precocious puberty. However, because GnRH may not be commercially available for testing purposes in several countries, most stimulation tests are now carried out using a GnRH agonist (GnRHa), such as leuprolide acetate or naferelin. After administration of leuprolide acetate, LH levels are measured after 1 and 2 hours.
Measurement of Gonadotropins in the Diagnosis of Puberty Alterations
Using ultrasensitive LH assays, measurement of basal serum LH levels is often sufficient for making the diagnosis of central precocious puberty (CPP) while stimulation tests are reserved for those patients with inconclusive basal results. Some studies have reported that, using a third generation IMA, blood values of LH higher than 0.8 mIU/mL are sufficient for the diagnosis of gonadotropin-dependent precocious puberty. However, the sensitivity of the basal LH level for the diagnosis of CPP in girls is relatively low (about 65%) and about one third of patients with CPP have undetectable LH levels. Therefore while an increased basal LH level is highly suggestive of the diagnosis, a low value does not exclude a central pubertal activation.
In patients with signs of precocious puberty but inconclusive basal LH levels, a GnRHa test is usually carried out. The test may be performed in many ways but most centers use leuprolide acetate at a dose of 20 µg/kg, up to a maximum of 500 µg. Peak LH of at least 5 IU/L indicates a pubertal response.
In most instances, gonadotropin measurements in the basal state and after the GnRHa test are not able to distinguish delayed puberty from hypogonadotropic hypogonadism.
Measurement of Gonadotropins in Adult Female Reproductive Disorders
High values of LH with increased LH:FSH ratios are found in 50% of women with PCOS. In the past, some clinicians based the diagnosis of PCOS on the finding of elevated LH and LH:FSH ratios higher than 2.0 or 3.0. While finding an increased serum LH or LH:FSH ratio may be useful for confirming the diagnosis of PCOS, many patients with PCOS have normal serum LH and LH:FSH ratios.
High levels of serum LH and FSH are typical of premature ovarian failure/insufficiency and may be found in patients with gonadal dysgenesis. Rarely, pituitary tumors may produce FSH or LH. Measurement of α-subunits and determination of the gonadotropin responses to thyrotropin-releasing hormone (TRH) may be useful for the diagnosis of gonadotropin adenomas.
Measurement of Prolactin
Serum prolactin (PRL) is generally measured by RIA or IRMA. The upper limit of the normal range is generally reported to be 15 to 20 ng/mL in men and 20 to 25 ng/mL in women, although the true normal levels are usually lower (up to 18 ng/mL in women). Serum PRL values are influenced by estrogens, drugs (e.g., phenothiazines, metoclopramide), stress, food consumption, breast stimulation, and even venipuncture. Because of diurnal changes and transient increases after meals, routine samples should be obtained at mid-morning.
In normal women, blood immunoreactive prolactin results predominantly from monomeric prolactin (≥95%). Dimeric prolactin forms (so called big-prolactin), and big-big prolactin, a macroprolactin (due to a prolactin-antibody complex of molecular weight >100 kDa), are generally present in low concentrations (<1%). However, macroprolactinemia is present in 15% to 35% of subjects having increased immunoreactive prolactin, and in some patients hyperprolactinemia is the result of elevated macroprolactin levels (without a commensurate biological effect) and often leads to a misleading medical approach.
The gold standard method for detecting macroprolactinemia is by gel filtration chromatography, a procedure that allows for quantification of all three variants of PRL. However, this method is labor intensive and most clinical laboratories prefer using precipitation with polyethylene glycol (PEG). A low PRL recovery after PEG treatment indicates the presence of macroprolactin.
Serum PRL levels greater than 150 ng/mL are diagnostic of the presence of a pituitary adenoma, but patients with microadenomas may have levels that are considerably lower.
Mild elevations of serum PRL may occur with certain CNS tumors or granulomas compressing the pituitary stalk. A mild elevation may also occur in hypothyroidism, with drugs stimulating PRL secretion, with high estrogen levels, and also during stress. If drugs and hypothyroidism are ruled out, magnetic resonance imaging (MRI) of the hypothalamic-pituitary region of the brain should be obtained. If no cause of hyperprolactinemia is found, macroprolactinemia should be ruled out, although this situation may be suspected when there are no symptoms or signs of elevated PRL, such as having normal menstrual function.
While it has been suggested to screen for macroprolactin in all hyperprolactinemic patients, since the Pituitary Society has published guidelines indicating that values of blood prolactin ≥150 ng/mL are always suggestive of pituitary prolactinoma, we suggest screening for macroprolactin only in patients having serum PRL values between 25 and 150 ng/mL. PRL recovery ≤30% following PEG precipitation indicates the presence of macroprolactinemia. Because true hyperprolactinemia and macroprolactinemia may coexist, post PEG PRL values should not exceed 13 ng/mL.
In pregnancy, serum PRL begins to increase by 6 weeks gestation and rises progressively to reach approximately 200 ng/mL at term, although the variability is large. In nonlactating women, PRL levels return to normal 2 to 3 weeks postpartum. With menopause, serum PRL declines slightly as a result of the reduction of estrogen.
Measurement of Estradiol and Other Estrogens
In most laboratories using commercial immunoassays, serum estradiol ranges between 20 and 80 pg/mL during the early to mid-follicular phases of the menstrual cycle and peaks at 200 to 500 pg/mL during the preovulatory LH surge. Refinements in assays, as discussed earlier and later, now show that these general “normal” ranges are higher than true values. However, most commercial assays and kits are not able to measure the low levels of estradiol that may be found in children, men, postmenopausal women, and women receiving aromatase inhibitors for the treatment of breast cancer. Before puberty, serum estradiol levels are under 20 pg/mL, indistinguishable from cases of hypogonadism. After menopause, estradiol levels fall to prepubertal levels; mean serum estradiol is typically between 10 and 20 pg/mL, and levels are lower than 10 pg/mL in women who have undergone oophorectomy. Similar problems in the estradiol assay exist in men where serum estradiol should be less than 40 pg/mL.
The advent of radioimmunometric and other immunoassay methods has not improved the precision and accuracy of estradiol assays. For measuring low levels, samples should be extracted with organic solvents and subjected to chromatographic separation to remove interfering steroids, particularly estrogen conjugates. Recently, simpler analytical methods using LC coupled with MS have been developed and are increasingly used for measuring E2 samples. These methods are particularly useful when a low estradiol level is expected and has a sensitivity, which is adequate to monitor patients with breast cancer treated with aromatase inhibitors. In these patients, methods need to be able to distinguish between suppressed levels of 1 pg/mL or less, from the pretreatment levels that are commonly 10 to 15 pg/mL.
The measurement of serum estradiol in men may be useful in the clinical evaluation of gynecomastia. In pubertal gynecomastia, a normal estradiol level is generally found. The finding of increased estradiol, testosterone, and LH but normal FSH in subjects with pubertal gynecomastia may indicate a syndrome of partial androgen insensitivity.
Serum estrone is higher than values of estradiol in women after menopause and in anovulatory women who do not produce adequate amounts of estradiol via the dominant follicle. Estrone levels are increased with obesity and are most closely linked to the increase in adipose tissue, where aromatization is increased. In women receiving any oral estrogen, estrone levels exceed those of estradiol; and its measurement is sometimes useful for the assessment of absorption and metabolism problems with oral estrogen therapy. Although not frequently measured, estrone sulfate reflects the circulating reservoir of estrogen and is quantitatively the highest circulating estrogen.
Estriol may be measured by RIA in serum or by spectrophotometric methods or RIA in urine. Estriol, which is produced in the fetal-placental unit, may be analyzed for prenatal genetic testing to help detect aneuploidy, but is not useful today for the assessment of fetal well-being.
At present there is no clinical utility for the measurements of estetrol or catecholestrogens.
Measurement of Progesterone
Serum progesterone may be measured by a variety of immunoassays. The specificity of the assay is highly dependent on lack of cross reactivity with several pregnane metabolites.
Blood Levels of Progesterone and the Assessment of Ovulatory Function
Serum progesterone is low during the follicular phase, with levels being less than 1.5 ng/mL. Levels begin to increase just before the onset of the LH surge and then increase progressively to peak 6 to 8 days after ovulation. After menopause, serum progesterone of adrenal origin is under 0.5 ng/mL.
The measurement of serum progesterone during the mid-luteal phase (days 21 and 22) is most frequently used to assess ovulatory status. Although single samples are acceptable in clinical practice, it should be appreciated that progesterone levels exhibit a pulsatile pattern as well as some diurnal variation.
During the mid-luteal phase, serum progesterone levels are usually higher than 7 ng/mL. Some physicians have proposed using three luteal determinations with a total serum value of 15 ng/mL or more to indicate normal luteal function. For patients being monitored for fertility, other assessments may be used including basal body temperature charts, urinary LH kits, and timed endometrial biopsies. In conception cycles, properly timed mid-luteal progesterone levels are over 10 ng/mL. Progesterone is also often used to assess ovulation after induction of ovulation. In clomiphene cycles, mid-luteal progesterone levels should be over 15 ng/mL. In assisted reproduction cycles such as in vitro fertilization (IVF) where gonadotropins are administered, a small rise in progesterone (>2 ng/mL) has been linked to decreased pregnancy rates. This is related to luteinization rather than premature ovulation per se, but this level is detrimental to endometrial receptivity.
An alternative to the measurement of serum progesterone is urinary pregnandiol glucuronide. This provides a more integrated assessment of luteal function and is usually normalized for urinary excretion by the determination of creatinine.
During pregnancy, serum progesterone may be useful to assess corpus luteum and placental function. Maternal serum progesterone levels nadir in early pregnancy at 9 weeks with levels of approximately 10 ng/mL and then rise slowly to 40 ng/mL near the end of the first trimester and then increase progressively to reach 150 ng/mL at term. Low levels of progesterone (below 10 ng/mL) at 6 to 8 weeks signify an abnormal intrauterine pregnancy or an ectopic pregnancy.
Measurement of Androgens
Androgens in women arise from three different sources: the ovaries, the adrenal glands, and the peripheral compartment. Most androgens are produced or metabolized by more than one compartment and, in evaluating women with androgen excess, several androgens are usually measured. In the past, the measurement of urinary 17-ketosteroids (17-KS) was the most common method used to evaluate androgen production in women. However, 17-KS measurements reflect adrenal androgen production, poorly reflect testosterone production, and are nonspecific; therefore they have been abandoned for use in current practice.
Most commonly, serum testosterone and dehydroepiandrosterone sulfate (DHEAS) are measured, which largely reflect ovarian and adrenal contributions, respectively. Serum testosterone reflects mostly ovarian androgen production, with two thirds of circulating levels resulting from the peripheral conversion of androstenedione; therefore, increases in either ovarian or adrenal androstenedione production results in elevated serum testosterone levels. On the other hand, while serum DHEAS reflects adrenal androgen secretion well, levels may be normal in certain cases of adrenal androgen hypersecretion (as in 21-hydroxylase deficiency). The pathway of adrenal enzymatic blockade (affecting the Δ 4 pathway) explains this discrepancy. Conversely, serum DHEAS may be elevated in many patients with a prevalent ovarian source of hyperandrogenemia (as in PCOS) and this is explained by the peripheral conversion of ovarian-derived DHEA to DHEAS in the circulation. Similarly, the increased levels of serum DHEAS also do not predict androgen responses to dexamethasone suppression, which has been used as a test to determine adrenal responsivity to suppression.
Serum testosterone should be measured by RIA or ICMA, after extraction and chromatography. These assays are cumbersome, time-consuming, and relatively costly, and accordingly many laboratories use direct assays without purification. However, these methods have some major disadvantages including overestimation of the values and low specificity. It has been reported that these direct testosterone assays have poor or no validity in females.
Many commercial diagnostic laboratories have switched to MS after liquid chromatography (LC/MS) assays. It has been suggested that this method has high sensitivity and specificity, and permits a better differentiation between normal and increased levels in women. However, this method does not provide better results than those obtained with classic methods of testosterone evaluation that include extraction.
Unbound testosterone may be measured as bioavailable testosterone or as free testosterone. The first method, which is also referred to as non-SHBG-bound testosterone, relies on SHBG to assess the percentage that is “free.” In healthy women, up to 75% of testosterone is bound to SHBG; the percentage that is unbound includes testosterone that is entirely “free” and the moiety associated with albumin that is available to the target cells.
Free testosterone is measured by equilibrium dialysis and has remained the gold standard for assessing testosterone that is neither bound to SHBG nor associated with albumin. Variations in incubation temperature significantly affect results in the dialysis method, which renders this method, which is tedious and expensive, rarely used clinically.
Commercial methods measure unbound testosterone by a direct method using a 125 I-labeled testosterone analogue as tracer. This analogue method gives 75% lower values than equilibrium dialysis assays and, although it is easy to perform, its clinical utility is questionable. It has been reported that direct assays of free testosterone result in unacceptably spurious values with high random variability and, therefore, should not be used in clinical practice. It is most practical for clinical purposes, to calculate a free testosterone index (free androgen index, FAI) using levels of testosterone and SHBG and albumin. An average normal albumin concentration from the literature can be used in the equation, and the binding constants. Calculated free testosterone levels in women have been found to be nearly identical with corresponding values determined by equilibrium dialysis. The calculation is made using the ratio between testosterone and SHBG (in units of nmol/L): (T (ng/mL) × 3.467/SHBG × 100). However, FAI values have low validity in adult males and are strongly dependent on the accuracy of testosterone and SHBG assays. In general, FAI should be used primarily for clinical rather than research purposes, when testosterone assays using chromatography and purification techniques are not available. It has also been thought that since saliva is a natural distillate, measurements of salivary testosterone may provide a measure of “unbound” testosterone. Lack of precision and reliability with salivary testosterone do not allow this measurement to be recommended for clinical use.
There are other androgens that may be useful to measure under different circumstances. These include androstenedione and 11β-hydroxyandrostenedione, the latter of which reflects adrenal production of androstenedione, because 11β-hydroxylase activity is usually absent in the ovary.
Assessment of peripheral androgen production requires the measurement of products of 5α-reductase activity. While serum dihydrotestosterone (DHT) does not appropriately reflect increases in peripheral 5α-reductase activity, more distal metabolites may be helpful. Serum 3α-androstanediol glucuronide best reflects peripheral testosterone metabolism in hirsutism, and androsterone glucuronide best reflects the state in androgenic acne.
In assessing androgen excess in women, it is preferable to obtain blood samples in the morning because of diurnal variation from the adrenal. Values are often reduced during the afternoon or evening.
Blood Levels of Androgens in Women
In women of reproductive age, an accurate range for serum testosterone is between 20 and 50 ng/dL. With direct immunoassays, the upper end of the normal range is approximately 70 to 80 ng/dL, or even higher. Serum testosterone levels higher than 200 ng/dL are suggestive of ovarian neoplasms, but some tumors may present with lower levels. Because upper normal ranges vary, a rule of thumb is to be concerned about a tumor in women when values exceed 2.5 times the upper normal range of a particular assay. Patients with PCOS have mildly increased serum testosterone (usually to values <100 ng/dL), while markedly increased levels may be found in patients with hyperthecosis and in those who have classic congenital adrenal enzymatic deficiencies. Patients with nonclassical congenital adrenal hyperplasia (NCAH) have androgen values similar to those found in PCOS.
The normal range of serum DHEAS is between 0.5 and 2.8 µg/mL. Values above 8 µg/mL are suggestive of an adrenal neoplasm; if the adrenal tumor produces testosterone, however, only modestly elevated values (3 to 4 µg/mL) may be found.
Serum androstenedione levels range between 1.0 and 2.5 ng/mL, and the highest levels occur with a functioning tumor or in congenital adrenal hyperplasia.
The reference ranges for free and bioavailable testosterone can vary considerably depending on the assay method used to determine these testosterone fractions. Again, it is important to know the reference ranges in the specific laboratory where the samples are being analyzed. Reference ranges of 1.1 to 14.3 ng/dL and 1.1 to 6.3 ng/mL for bioavailable and free testosterone, respectively, are used in one large clinical diagnostic laboratory. The reference range for SHBG in women is generally approximately 30 to 90 nmol/L.
Serum androgens are low before puberty, with adrenal androgen levels beginning to increase 1 or 2 years before the onset of the puberty. Serum DHEAS values higher than 0.8 µg/mL indicate the presence of adrenarche.
In women, adrenal androgens begin to decline in the third decade of life and the greatest reduction in values occurs between age 20 and the fifth and sixth decades of life. Ovarian androgens begin to decline a little later, generally in the third decade and this reduction continues after menopause. Similar declines of ovarian and adrenal androgens are observed in PCOS and may explain the improvement in menstrual function observed in many patients with PCOS after age 40.
Measurement of serum DHEA-S levels may be helpful in that very low levels for age may suggest adrenal deficiency as occurs in young women with autoimmune ovarian failure.
Measurement of Circulating Androgen Precursors in Women
Measurements of intermediates in steroid metabolism are useful for the diagnosis of adrenal enzymatic deficiencies. Serum 17α-OHP is used in the diagnosis of 21-hydroxylase deficiency. Because 17α-OHP is secreted by the corpus luteum, the measurement should be carried out in the morning during the follicular phase. In healthy women, serum 17α-OHP is generally lower than 1 ng/mL, but hyperandrogenic patients (mostly those with PCOS) generally have slightly higher levels.
Patients with classic neonatal forms of 21-hydroxylase deficiency have very high levels of 17α-OHP (50 to 200 ng/mL), while the diagnosis of NCAH relies mostly on the finding of serum 17-OHP concentrations above 10 ng/mL (30 nmol/L). An increased basal 17-OHP result between 2 and 10 ng/mL (30 nmol/L) must be confirmed by the finding of 17-OHP above 10 ng/mL (30 nmol/L) after stimulation of adrenal function by the administration of synthetic ACTH (cosyntropin), which is usually given as an intravenous bolus but can also be given intramuscularly.
From a practical standpoint, the CYP21A2 locus responsible for 21-hydroxylase deficiency is complex, precluding its molecular genetic analysis as the first line diagnostic test for NCAH. However, it is essential for genetic counseling since many patients with NCAH carry a severe allele, which may result in findings associated with CAH in the progeny.
The measurement of serum 11-deoxycortisol may be used to diagnose 11β-hydroxylase deficiency when 17α-OHP is elevated. This is a rarer condition that may be associated with hypertension.
The measurement of serum 17α-hydroxypregnenolone (and the ratio of 17α-OHP to 17α-hydroxypregnenolone), as well as the ratio for DHEA to androstenedione is useful in the diagnosis of 3β-hydroxysteroid dehydrogenase deficiency. In adults, no genetic mutations in the 3βHSD gene have been uncovered and most patients previously diagnosed with this enzymatic deficiency probably have PCOS with an enhanced adrenal component.
Adrenocorticotropic Hormone Stimulation Test
Because serum androgens may arise from different sources in women, several tests have been established to distinguish adrenal from ovarian hyperandrogenism. However, most tests are not specific and their use is generally limited. Only the ACTH stimulation test is currently used.
The ACTH stimulation test is generally used for the diagnosis of cortisol deficiency or to uncover mild adrenal enzymatic deficiencies such as CAH. Also, in the past it has been used to distinguish between adrenal and ovarian sources of hyperandrogenism, but is not generally used for this purpose because of its lack of specificity.
The test is best performed between 8 and 9 a.m., with 0.25 mg of cosyntropin injected intravenously. While the test may also be accomplished by intramuscular injection, more consistent results have been obtained with intravenous administration. Blood samples are generally obtained at 30 and 60 minutes, but the 60-minute value suffices as a single time point for practical purposes. For the diagnosis of nonclassic 21-hydroxylase deficiency, serum cortisol and 17α-OHP are measured. Serum 11-deoxycortisol may be evaluated if the rare 11-hydroxylase defect is suspected. Nonclassic 21-hydroxylase deficiency may be diagnosed if peak 17α-OHP is higher than 10 ng/mL 2 ( Fig. 34.5 ).
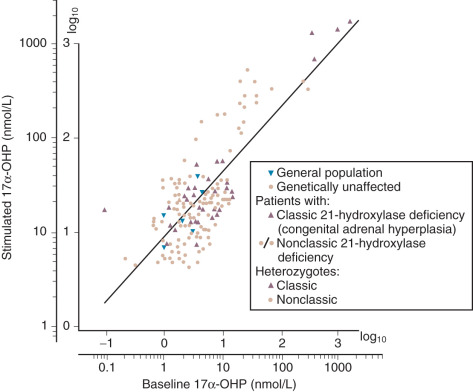
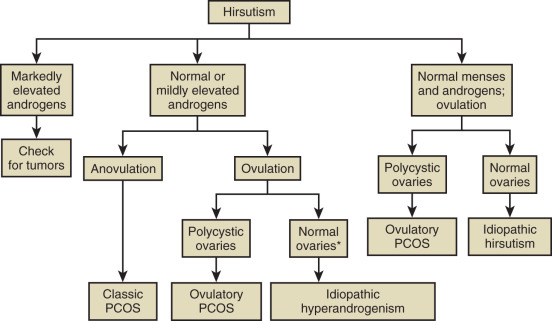
Hormonal Evaluation of Hirsutism
Hirsutism is excessive hair growth in women where it is not normally found, usually with a central body distribution. When this biologic signal is manifest, laboratory tests may be indicated to pinpoint the abnormality. Most commonly, total testosterone, DHEAS, and SHBG are measured (as well as 17α-OHP to exclude nonclassic congenital adrenal hyperplasia). In about 10% to 15% of hirsute women, all of these hormone levels are in the normal range and with the occurrence of normal menstrual cycles, the diagnosis of “idiopathic hirsutism” is made. While it is not our intent to discuss details of the differential diagnosis of hirsutism, a simple algorithm is provided in Fig. 34.6 that describes the possible diagnoses. There are several other algorithms for this evaluation, namely that of the Androgen Excess and PCOS Society and a recent updated guideline by the Endocrine Society. Specifically the latter guideline stresses that only the measurement of testosterone is needed initially, with other measurements being reserved for when there is a suspicion for other disorders such as PCOS or NCAH.
Related posts:
 Gonadotropin Hormones and Their Receptors
Gonadotropin Hormones and Their Receptors
 Steroid Hormones and Other Lipid Molecules Involved in Human Reproduction
Steroid Hormones and Other Lipid Molecules Involved in Human Reproduction
 Structure, Function, and Evaluation of the Female Reproductive Tract
Structure, Function, and Evaluation of the Female Reproductive Tract
 Endocrine Disturbances Affecting Reproduction
Endocrine Disturbances Affecting Reproduction
 Physiologic and Pathophysiologic Alterations of the Neuroendocrine Components of the Reproductive Axis
Physiologic and Pathophysiologic Alterations of the Neuroendocrine Components of the Reproductive Axis
 Pelvic Imaging in Reproductive Endocrinology
Pelvic Imaging in Reproductive Endocrinology
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


