FIGURE 16.1 Progression of stages in esophageal squamous cell cancer and esophageal adenocarcinoma.
Cyclin D1 and p16INK4a
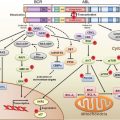
The mammalian cell cycle is regulated exquisitely by cyclins, cyclin-dependent kinases (CDK), and cyclin-dependent kinase inhibitors (CDKi such as p15, p16, p21, and p27). During G1 phase, the cyclin D1 oncogene complexes with either CDK4 or CDK6 to phosphorylate the retinoblastoma (pRb) tumor suppressor protein and, in so doing, relieves the negative regulatory effect of pRb, allowing the E2F family of transcription factors to propel the cell cycle toward the G1/S phase transition.16 Toward the late G1 phase, cyclin E complexes with CDKs to phosphorylate p107, which is related to pRb, and liberate more E2F members to navigate the cell cycle into S phase. As with EGFR, cyclin D1 overexpression is found in premalignant lesions, such as esophageal squamous dysplasia or Barrett’s esophagus, and the majority of early-stage ESCC or EAD.17,18 Additionally, cyclin D1 overexpression correlates with poor outcomes and survival as well as poor response to chemotherapy.19,20
TABLE 16.1
COMMON MOLECULAR GENETIC ALTERATIONS OBSERVED IN ESOPHAGEAL AND GASTRIC CANCERS

Although cyclin D1 overexpression accounts for cyclin D1 dysregulation, other mechanisms include mutations in cyclin D1 and mutations in Fbx4, which is the E3 ligase for cyclin D1, thereby preventing degradation of cyclin D1 in the cytoplasm and reimportation into the nucleus, where it exerts its oncogenic effects.21
In a similar vein, p16INK4a is an early genetic alteration, via promoter hypermethylation, point mutation, or allelic deletion, in Barrett’s esophagus and EAD, but interestingly, a late event in ESCC. Loss of heterozygosity of 9p21, the locus for both p16 and p15, has been demonstrated with high frequency in both dysplastic Barrett’s epithelium and Barrett’s adenocarcinoma (90% and more than 80% of cases, respectively).22,23 Promoter hypermethylation, which prevents tumor suppressor function by blocking transcription, has been documented and correlates with the degree of dysplasia in Barrett’s esophagus. It is present in up to 75% of specimens with high-grade dysplasia and is found in almost 50% of patients with adenocarcinoma of the esophagus.24 Point mutations of p16 in ESCC have been found and promoter hypermethylation has been noted in up to 50% of these tumors.25,26 Rb gene mutation is not found in either type of esophageal neoplasm, but allelic loss of 13q where the locus of the Rb gene resides is found in up to 50% of patients with Barrett’s adenocarcinoma and squamous cell carcinoma.18,27 This can correlate with diminished or loss of pRb protein in Barrett’s esophagus with dysplasia, EAD, and ESCC.28,29
p53 Tumor Suppressor Gene
p53 is one of the most commonly mutated genes in human cancer.22–24 p53 (molecular weight approximately 53 kDa) is a tumor suppressor that interrupts the G1 phase to evaluate and permit repair of damaged DNA, which may arise from environmental exposure (e.g., irradiation, ultraviolet light) or cellular stress.30 In the face of irreparable damage, p53 induces apoptosis. The p53 transcription factor binds DNA to activate or suppress a large repertoire of target genes.31 p53 mutations induce loss of cell-cycle checkpoints and promote genomic instability. The majority of p53 mutations occur in the DNA-binding region, and more than 80% of them are missense mutations resulting in loss of wild type p53 function.32 Wild type p53 has a short half-life and is difficult to detect by immunohistochemistry; mutation in p53 results in stabilization of the protein and allows for easier detection by immunohistochemistry.
Detection of mutated p53 protein by immunohistochemistry has been demonstrated with increasing frequency during histologic progression from Barrett’s esophagus (5%) through dysplasia (65% to 75%) to frank adenocarcinoma (up to 90%).33–36 Thus, p53 mutation or loss of heterozygosity appears early in Barrett’s esophagus and EAD. Both mutant p53 protein detected by immunohistochemistry and specific p53 gene mutations detected by genomic sequencing have been identified in 40% to 75% of patients with ESCC.37–40 The presence of a p53 point mutation correlates with response to induction chemoradiotherapy and predicted survival after esophagectomy in patients with either ESCC or EAD.41
Telomerase Activation
Maintenance of telomere length allows DNA replication to be sustained indefinitely. Aberrant expression of telomerase has been observed in most esophageal cancers examined to date.42 Morales et al.43 observed increased telomerase expression in 100% of adenocarcinoma and Barrett’s esophagus cases with high-grade dysplasia. Telomerase activation is important, but alternative mechanisms to maintain the length of telomeres may operate in these cancers as well.44
Tumor Invasion and Metastasis
Loss of cell-cell adhesion can lead to both invasion and metastases. Alterations in expression of E-cadherin, a cell-cell adhesion molecule, or its associated catenins disrupt cell-cell interactions, which results in the potential for tumor progression.45 Reduced expression of E-cadherin has been correlated with progression from Barrett’s esophagus to dysplasia and finally to adenocarcinoma, and also observed in ESCC.46,47
Models of ESCC and EAD
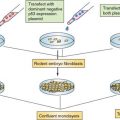
Advances in diagnosis and therapy of esophageal neoplasms will ultimately be fostered through cell lines, xenotransplantation mouse models, surgically based rodent models, and genetically engineered mouse models. There is a vast array of cell lines established from primary and metastatic human esophageal cancers that allow perturbation of gene expression to gauge effects on cellular behavior. Recently, organotypic (three-dimensional) cell culture models, which mimic human tissue, have revealed that the combination of EGFR and mutant p53 results in transformation of human esophageal epithelial cells immortalized with hTERT.48 A classic rodent model involves total gastrectomy followed by esophagojejunostomy.49 This creates a milieu whereby the esophagus is exposed to high concentrations of bile (“nonacid reflux”) with the development of Barrett’s esophagus and EAD. In transgenic mice in which cyclin D1 is targeted to the esophagus, esophagi reveal evidence of dysplasia that evolves into squamous cell cancer on crossbreeding the mice with p53 haploinsufficiency or loss.50 Rodents have also been treated with nitrosamines to yield esophageal papillomas and ESCC.51,52
The underlying fate switch between ESCC and EAD may be influenced as well by the expression and function of “lineage”-specific transcriptional factors as demonstrated through functional genomics. To that end, SOX2, found to be part of an amplicon on chromosome 3q26.33 in human ESCC, fosters growth of these cancers. This may have implications in the therapy of human ESCC.53
MOLECULAR BIOLOGY OF GASTRIC CANCER
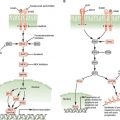
The most common type of gastric cancer is adenocarcinoma, of which there are two subtypes: intestinal and diffuse. They are distinguished by different anatomic locations within the stomach, variable clinical outcomes, and different pathogenesis. The intestinal type of sporadic gastric adenocarcinoma has a hallmark progression from normal gastric epithelium to chronic atrophic gastritis (typically due to Helicobacter pylori infection) to intestinal metaplasia (which has some overlapping but also different features than intestinal metaplasia of Barrett’s esophagus) to dysplasia to cancer (Fig. 16.2). Diffuse-type gastric adenocarcinoma is even more invasive and aggressive in its behavior, has overlap with lobular-type breast cancer, and may be highlighted by loss of E-cadherin.
Inherited Susceptibility
Case-control studies have observed consistent, up to threefold, increases in risk for gastric cancer among relatives of patients with gastric cancer.54,55 Studies of monozygotic twins have even shown a slight trend toward increased concordance of gastric cancers compared with dizygotic twins.56,57 Large families with an autosomal dominant, highly penetrant inherited predisposition for the development of gastric cancer are rare. However, early-onset diffuse gastric cancers have been described and linked to the E-cadherin/CDH1 locus on chromosome 16q and associated with mutations in this gene.58 This seminal finding has been confirmed in other studies with gastric cancers at a relatively high (67% to 83%) penetrant rate.59–62 Thus, E-cadherin mutation testing should be considered in the appropriate clinical setting. In fact, prophylactic gastrectomy should be considered strongly in families with germ line E-cadherin mutation even without gross mucosal abnormalities by endoscopic examination of the stomach.63

FIGURE 16.2 Progression of stages in intestinal-type gastric adenocarcinoma.
Lynch syndrome or hereditary nonpolyposis colon cancer involves germ line mutations of DNA mismatch repair genes.64 Gastric adenocarcinoma may be observed in some families with Lynch syndrome. Gastric cancers have also been noted to occur in patients with familial adenomatous polyposis and Peutz-Jeghers syndrome.
The Role of Helicobacter pylori Infection and Other Host-Environmental Factors
As a commensal organism, H. pylori infection is widely prevalent throughout the world. Despite its classification by the World Health Organization as a class I carcinogen, infection with H. pylori does not typically lead to gastric cancer. This underscores the importance of other factors, such as virulence, environmental, and host factors, as well as genetic polymorphisms (e.g., in interleukin-1β, a potent inhibitor of acid secretion).65 The blood group A phenotype has been reported to be associated with gastric cancers.66,67 H. pylori may adhere to the Lewis blood group antigen, indicating a factor for increased risk for gastric cancer.68 Small variant alleles of a mucin gene, Muc1, were found to be associated with gastric cancer patients when compared with a blood donor control population.69 Epstein-Barr virus infection has been noted in a certain type of gastric carcinoma (lymphoepithelioid type), although the importance of this is unclear.70
Molecular Genetic Alterations
In contrast to ESCC, EAD, pancreatic cancer, and colon cancer in which certain oncogenes and tumor suppressor genes are altered with high frequency, such degree of alteration is not observed in sporadic gastric cancers. A reasonably prevalent alteration is microsatellite instability, the result of changes in DNA mismatch repair genes (Table 16.1). Microsatellite instability and associated alterations of the TGF-beta II receptor, IGFRII, BAX, E2F-4, hMSH3, and hMSH6 genes are found in a subset of gastric carcinomas.71–75 Microsatellite instability has been found in 13% to 44% of sporadic gastric carcinomas.76 A high degree of microsatellite instability occurs in gastric cancers of the intestinal type, reduced involvement of lymph nodes, enhanced lymphoid infiltration, and better prognosis.77 This is reminiscent of colon cancers associated with Lynch syndrome.
The p53 tumor suppressor gene is consistently altered in most gastric cancers.78 In a study of the promoter region of p16 in gastric cancers, a significant number (41%) exhibited CpG island methylation.79 Many cases with hypermethylation of promoter regions displayed the phenotype with a high degree of microsatellite instability and multiple sites of methylation, including the hMLH1 promoter region.80
Many sporadic diffuse gastric cancers display altered E-cadherin, a transmembrane, calcium-dependent adhesion molecule important in epithelial cell homophilic and heterophilic interactions. E-cadherin may be down-regulated in gastric carcinogenesis (especially diffuse gastric adenocarcinoma) by point mutation, allelic deletion, or promoter methylation.81,82 In addition, during epithelial-mesenchymal transition, E-cadherin transcription can be silenced by transcriptional factors such as Snail and Slug. However, it is not clear if epithelial-mesenchymal transition is an important process in gastric carcinogenesis, as is believed to be the case, for example, in breast cancer.
Alterations in a number of other oncogenes and tumor suppressor genes have been described in a very small subset of gastric cancers by polymerase chain reaction–based or immunohistochemical analysis, but the variability in methods and lack of uniformity in quality control make these observations less compelling. As with esophageal cancer, high-throughput assays, such as single-nucleotide polymorphism arrays, chromosomal genomic hybridization (to assess chromosomal gains and losses) arrays, gene expression profiling through microarrays, and tissue- and plasma-based proteomics may unravel molecular signatures (and even specific genes and/or pathways) that define subtypes of gastric cancers, different stages of gastric cancers, and correlations with clinical outcomes.
Models of Gastric Cancer
Genetically engineered mouse models of gastric cancer have emerged in rapid fashion in recent years, indicating that activated Wnt signaling and induced downstream effectors, p53 inactivation, APC gene inactivation, Smad4 gene inactivation, and gastrin are critical factors.83–87 Gastric cancers in these protean mouse models are facilitated by concomitant infection with Helicobacter.88,89 Furthermore, recruitment of bone marrow stem cells may augment the effects of Helicobacter infection during gastric carcinogenesis.90
Recently, it has been demonstrated that overexpression of interleukin-1β in mice results in gastric inflammation and cancer, with concomitant recruitment of immature myeloid cells (also referred to as myeloid-derived suppressor cells).91
Selected References
The full list of references for this chapter appears in the online version.
1. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000;100:57.
3. Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell 2000;103(2): 211.
5. Torzewski M, Sarbia M, Verreet P, et al. The prognostic significance of epidermal growth factor receptor expression in squamous cell carcinomas of the oesophagus. Anticancer Res 1997;17(5B):3915.
8. Jankowski J, McMenemin R, Hopwood D, et al. Abnormal expression of growth regulatory factors in Barrett’s oesophagus. Clin Sci (Lond) 1991;81: 663.
10. Jankowski J, Hopwood D, Wormsley KG. Flow-cytometric analysis of growth-regulatory peptides and their receptors in Barrett’s oesophagus and oesophageal adenocarcinoma. Scand J Gastroenterol 1992;27:147.
12. Yacoub L, Goldman H, Odze RD. Transforming growth factor-alpha, epidermal growth factor receptor, and MiB-1 expression in Barrett’s-associated neoplasia: correlation with prognosis. Mod Pathol 1997;10:105.
13. Itakura Y, Sasano H, Shiga C, et al. Epidermal growth factor receptor overexpression in esophageal carcinoma: an immunohistochemical study correlated with clinicopathologic findings and DNA amplification. Cancer 1994; 74:795.
17. Arber N, Lightdale C, Rotterdam H, et al. Increased expression of the cyclin D1 gene in Barrett’s esophagus. Cancer Epidemiol Biomarkers Prev 1996;5:457.
19. Shamma A, Doki Y, Shiozaki H, et al. Cyclin D1 overexpression in esophageal dysplasia: a possible biomarker for carcinogenesis of esophageal squamous cell carcinoma. Int J Oncol 2000;16:261.
20. Sarbia M, Bektas N, Muller W, et al. Expression of cyclin E in dysplasia, carcinoma, and nonmalignant lesions of Barrett esophagus. Cancer 1999;86: 2597.
21. Barbash O, Zamfirova P, Lin DI, et al. Mutations in Fbx4 inhibit dimerization of the SCF(Fbx4) ligase and contribute to cyclin D1 overexpression in human cancer. Cancer Cell 2008;14(1):68.
22. Barrett MT, Sanchez CA, Galipeau PC, et al. Allelic loss of 9p21 and mutation of the CDKN2/p16 gene develop as early lesions during neoplastic progression in Barrett’s esophagus. Oncogene 1996;13:1867.
24. Klump B, Hsieh CJ, Holzmann K, et al. Hypermethylation of the CDKN2/p16 promoter during neoplastic progression in Barrett’s esophagus. Gastroenterology 1998;115:1381.
25. Xing EP, Nie Y, Wang LD, et al. Aberrant methylation of p16INK4a and deletion of p15INK4b are frequent events in human esophageal cancer in Linxian, China. Carcinogenesis 1999;20:77.
26. Maesawa C, Tamura G, Nishizuka S, et al. Inactivation of the CDKN2 gene by homozygous deletion and de novo methylation is associated with advanced stage esophageal squamous cell carcinoma. Cancer Res 1996;56:3875.
28. Coppola D, Schreiber RH, Mora L, et al. Significance of Fas and retinoblastoma protein expression during the progression of Barrett’s metaplasia to adenocarcinoma. Ann Surg Oncol 1999;6:298.
29. Ikeguchi M, Oka S, Gomyo Y, et al. Clinical significance of retinoblastoma protein (pRB) expression in esophageal squamous cell carcinoma. J Surg Oncol 2000;73:104.
33. Hamelin R, Flejou JF, Muzeau F, et al. TP53 gene mutations and p53 protein immunoreactivity in malignant and premalignant Barrett’s esophagus. Gastroenterology 1994;107:1012.
35. Younes M, Lebovitz RM, Lechago LV, et al. p53 protein accumulation in Barrett’s metaplasia, dysplasia, and carcinoma: a follow-up study. Gastroenterology 1993;105:1637.
37. Gaur D, Arora S, Mathur M, et al. High prevalence of p53 gene alterations and protein overexpression in human esophageal cancer: correlation with dietary risk factors in India. Clin Cancer Res 1997;3:2129.
38. Kato H, Yoshikawa M, Miyazaki T, et al. Expression of p53 protein related to smoking and alcoholic beverage drinking habits in patients with esophageal cancers. Cancer Lett 2001;167:65.
40. Taniere P, Martel-Planche G, Saurin JC, et al. TP53 mutations, amplification of P63 and expression of cell cycle proteins in squamous cell carcinoma of the oesophagus from a low incidence area in Western Europe. Br J Cancer 2001;85:721.
41. Ribeiro U Jr, Finkelstein SD, Safatle-Ribeiro AV, et al. p53 sequence analysis predicts treatment response and outcome of patients with esophageal carcinoma. Cancer 1998;83:7.
43. Morales CP, Lee EL, Shay JW. In situ hybridization for the detection of telomerase RNA in the progression from Barrett’s esophagus to esophageal adenocarcinoma. Cancer 1998;83:652.
44. Opitz OG, Suliman Y, Hahn WC, et al. Cyclin D1 overexpression and p53 inactivation immortalize primary oral keratinocytes by a telomerase-independent mechanism. J Clin Invest 2001;108(5):725.
45. Christofori G, Semb H. The role of the cell-adhesion molecule E-cadherin as a tumour-suppressor gene. Trends Biochem Sci 1999;24:73.
47. Takeno S, Noguchi T, Fumoto S, et al. E-cadherin expression in patients with esophageal squamous cell carcinoma: promoter hypermethylation, Snail overexpression, and clinicopathologic implications. Am J Clin Pathol 2004;122(1):78.
48. Okawa T, Michaylira CZ, Kalabis J, et al. The functional interplay between EGFR overexpression, hTERT activation, and p53 mutation in esophageal epithelial cells with activation of stromal fibroblasts induces tumor development, invasion, and differentiation. Genes Dev 2007;21(21):2788.
50. Opitz OG, Harada H, Suliman Y, et al. A mouse model of human oral-esophageal cancer. J Clin Invest 2002;110:761.
51. Siglin JC, Khare L, Stoner GD. Evaluation of dose and treatment duration on the esophageal tumorigenicity of N-nitrosomethylbenzylamine in rats. Carcinogenesis 1995;16(2):259.
53. Bass AJ, Watanabe H, Mermel CH, et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet 2009;41(11):1238.
58. Guilford P, Hopkins J, Harraway J, et al. E-cadherin germline mutations in familial gastric cancer. Nature 1998; 392:402.
59. Gayther SA, Gorringe KL, Ramus SJ, et al. Identification of germ-line E-cadherin mutations in gastric cancer families of European origin. Cancer Res 1998;58:4086.
62. Pharoah PD, Caldas C. Incidence of gastric cancer and breast cancer in CDH1 (E-cadherin) mutation carriers from hereditary diffuse gastric cancer families. Gastroenterology 2001;121:1348.
63. Lewis FR, Mellinger JD, Hayashi A, et al. Prophylactic total gastrectomy for familial gastric cancer. Surgery 2001;130(4):612.
64. Chung DC, Rustgi AK. The hereditary nonpolyposis colorectal cancer syndrome: genetics and clinical implications. Ann Intern Med 2003;138(7):560.
65. El Omar EM, Rabkin CS, Gammon MD, et al. Increased risk of noncardiac gastric cancer associated with proinflammatory cytokine gene polymorphisms. Gastroenterology 2003;124:1193.
69. Silva F, Carvalho F, Peixoto A, et al. MUC1 polymorphism confers increased risk for intestinal metaplasia in a Colombian population with chronic gastritis. Eur J Hum Genet 2003;11(5):380.
70. Lee HS, Chang MS, Yang HK, Lee BL, Kim WH. Epstein-Barr virus-positive gastric carcinoma has a distinct protein expression profile in comparison with Epstein-Barr virus-negative carcinoma. Clin Cancer Res 2004;10(5): 1698.
71. Kim SJ, Bang YJ, Park JG, et al. Genetic changes in the transforming growth factor beta (TGF-beta) type II receptor gene in human gastric cancer cells: correlation with sensitivity to growth inhibition by TGF-beta. Proc Natl Acad Sci U S A 1994;91:8772.
72. Yamamoto H, Sawai H, Perucho M. Frameshift somatic mutations in gastrointestinal cancer of the microsatellite mutator phenotype. Cancer Res 1997;57:4420.
76. Seruca R, Santos NR, David L, et al. Sporadic gastric carcinomas with microsatellite instability display a particular clinicopathologic profile. Int J Cancer 1995;64:32.
77. dos Santos NR, Seruca R, Constancia M, et al. Microsatellite instability at multiple loci in gastric carcinoma: clinicopathologic implications and prognosis. Gastroenterology 1996;110:38.
80. Toyota M, Ahuja N, Suzuki H, et al. Aberrant methylation in gastric cancer associated with the CpG island methylator phenotype. Cancer Res 1999;59:5438.
82. Grady WM, Willis J, Guilford PJ, et al. Methylation of the CDH1 promoter as the second genetic hit in hereditary diffuse gastric cancer. Nat Genet 2000;26:16.
83. Taketo MM. Wnt signaling and gastrointestinal tumorigenesis in mouse models. Oncogene 2006;25(57):7522.
84. Fox JG, Dangler CA, Whary MT, et al. Mice carrying a truncated Apc gene have diminished gastric epithelial proliferation, gastric inflammation, and humoral immunity in response to Helicobacter felis infection. Cancer Res 1997;57(18):3972.
85. Teng Y, Sun AN, Pan XC, et al. Synergistic function of Smad4 and PTEN in suppressing forestomach squamous cell carcinoma in the mouse. Cancer Res 2006;66(14):6972.
86. Watson SA, Grabowska AM, El-Zaatari M, Takhar A. Gastrin—active participant or bystander in gastric carcinogenesis? Nat Rev Cancer 2006;6(12): 936.
87. Wang TC, Dangler CA, Chen D, et al. Synergistic interaction between hypergastrinemia and Helicobacter infection in a mouse model of gastric cancer. Gastroenterology 2000;118(1):36.
88. Rogers AB, Taylor NS, Whary MT, et al. Helicobacter pylori but not high salt induces gastric intraepithelial neoplasia in B6129 mice. Cancer Res 2005;65(23):10709.
89. Cai X, Carlson J, Stoicov C, et al. Helicobacter felis eradication restores normal architecture and inhibits gastric cancer progression in C57BL/6 mice. Gastroenterology 2005;128(7):1937.
90. Houghton J, Stoicov C, Nomura S, et al. Gastric cancer originating from bone marrow-derived cells. Science 2004;306(5701):1568.
91. Tu S, Bhagat G, Cui G, et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 2008;14(5):408.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


