Esophagus and Stomach Cancers
Anil K. Rustgi
This chapter will deal with the molecular biology of esophageal and gastric cancers. There are several key aspects in the elucidation of the genetic basis of esophageal and gastric cancers through molecular biology approaches. These include, but are not limited to, new insights into underlying pathogenesis, possibilities for risk stratification and prognosis, correlations with traditional pathology classification schemes, development of new diagnostics, and potential applications in imaging and therapy. In considering the genetic underpinnings of esophageal and gastric cancers, or for any cancer, critical appraisal is required of oncogenes, tumor suppressor genes, and DNA mismatch repair genes as they modulate, either positively or negatively, growth factor receptor-mediating signaling cascades, transcription of target genes, and cell-cycle progression. These molecular networks conspire to influence cellular behaviors, such as proliferation, differentiation, apoptosis, senescence, and response to stress and injury. The exquisite equilibrium that is the signature of normal cellular homeostasis is perturbed in uncontrolled cell growth, resulting in eventual evolution of premalignant stages and malignant transformation. However, the time required for malignant transformation varies, depending on cellular- and tissue-specific context, and is affected by environmental factors.
The salient features of tumorigenesis and acquisition of the malignant phenotype that are required, as described by Hanahan and Weinberg,1 include growth signal autonomy, ability to surmount antigrowth signals, evasion of apoptosis, unlimited replicative ability, angiogenesis, and invasion and metastatic potential. More recently, the role of inflammation in carcinogenesis has gained much attention.
MOLECULAR BIOLOGY OF ESOPHAGEAL CANCER
The vast majority of esophageal cancers are of two subtypes: esophageal squamous cell cancer (ESCC) and esophageal adenocarcinoma (EAD). ESCC is preceded by squamous dysplasia, whereas EAD is preceded by Barrett’s esophagus or incomplete intestinal metaplasia of the normal squamous epithelium of the esophagus (Fig. 16.1). Barrett’s esophagus undergoes transition from low-grade and high-grade dysplasia before converting into EAD. ESCC and EAD have common and divergent genetic features as manifest by alterations in canonical oncogenes and tumor suppressor genes in somatic cells of tumors (Table 16.1). However, inherited predisposition to ESCC is rare, as described in tylosis palmaris et plantaris. Although the gene mutation for tylosis has remained elusive, the region of allelic deletion is on chromosome 17p.2 Similarly, there is no classic syndrome that distinguishes familial Barrett’s esophagus or familial EAD. That being said, studies continue to analyze families with Barrett’s esophagus in an effort to identify relevant genes or single-nucleotide polymorphisms. It is estimated that about 7% of patients with Barrett’s esophagus may have a family history. As a separate consideration, ESCC or EAD does not appear to emerge from infectious etiologies, although a small subset of ESCC is associated with human papillomavirus in some endemic regions of the world.
The Epidermal Growth Factor Receptor
The epidermal growth factor receptor (EGFR) family of receptor tyrosine kinases stimulates a number of signal transduction cascades (e.g., Ras/Raf/MEK/ERK, PI3K/AKT) that regulate diverse cellular processes, such as proliferation, differentiation, survival, migration and adhesion.3 These signaling pathways are important in normal cellular homeostasis, but aberrant activation of the EGFR members are crucial in esophageal carcinogenesis. This family of receptors comprises EGFR (also referred to as erbB1), erbB2, erbB3, and erbB4. The receptors have the ability to homo- or heterodimerize on engagement with one of several ligands: TGF (transforming growth factor)-α, EGF (epidermal growth factor), amphiregulin, heparinbinding EGF-like growth factor, betacellulin, and epiregulin. Tyrosine phosphorylation of homo- or heterodimers of EGFRs creates docking sites for
signaling proteins or adapter proteins. EGFR is commonly overexpressed in early-stage esophageal cancer, and overexpression correlates with a poor prognosis.4,5,6,7 EGFR overexpression is typically due to increased engagement with ligands and decreased turnover. However, mutation of a tyrosine residue in the cytoplasmic domain is rare. Increased expression of TGF-α and EGF has been detected in Barrett’s esophagus, EAD, and ESCC.8,9,10,11,12 EGFR overexpression may predict a poor response to chemoradiotherapy13,14 and is associated with decreased survival in patients with squamous cell carcinoma.13 Furthermore, EGFR overexpression was associated with recurrent disease and diminished overall survival in patients undergoing esophagectomy for ESCC.14,15 In contrast to EGFR, it is not clear if erbB2 overexpression is consistently found either in ESCC or EAD.
signaling proteins or adapter proteins. EGFR is commonly overexpressed in early-stage esophageal cancer, and overexpression correlates with a poor prognosis.4,5,6,7 EGFR overexpression is typically due to increased engagement with ligands and decreased turnover. However, mutation of a tyrosine residue in the cytoplasmic domain is rare. Increased expression of TGF-α and EGF has been detected in Barrett’s esophagus, EAD, and ESCC.8,9,10,11,12 EGFR overexpression may predict a poor response to chemoradiotherapy13,14 and is associated with decreased survival in patients with squamous cell carcinoma.13 Furthermore, EGFR overexpression was associated with recurrent disease and diminished overall survival in patients undergoing esophagectomy for ESCC.14,15 In contrast to EGFR, it is not clear if erbB2 overexpression is consistently found either in ESCC or EAD.
 FIGURE 16.1 Progression of stages in esophageal squamous cell cancer and esophageal adenocarcinoma. |
Cyclin D1 and p16INK4a
The mammalian cell cycle is regulated exquisitely by cyclins, cyclin-dependent kinases (CDK), and cyclin-dependent kinase inhibitors (CDKi such as p15, p16, p21, and p27). During G1 phase, the cyclin D1 oncogene complexes with either CDK4 or CDK6 to phosphorylate the retinoblastoma (pRb) tumor suppressor protein and, in so doing, relieves the negative regulatory effect of pRb, allowing the E2F family of transcription factors to propel the cell cycle toward the G1/S phase transition.16 Toward the late G1 phase, cyclin E complexes with CDKs to phosphorylate p107, which is related to pRb, and liberate more E2F members to navigate the cell cycle into S phase. As with EGFR, cyclin D1 overexpression is found in premalignant lesions, such as esophageal squamous dysplasia or Barrett’s esophagus, and the majority of early-stage ESCC or EAD.17,18 Additionally, cyclin D1 overexpression correlates with poor outcomes and survival as well as poor response to chemotherapy.19,20
TABLE 16.1 COMMON MOLECULAR GENETIC ALTERATIONS OBSERVED IN ESOPHAGEAL AND GASTRIC CANCERS | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
Although cyclin D1 overexpression accounts for cyclin D1 dysregulation, other mechanisms include mutations in cyclin D1 and mutations in Fbx4, which is the E3 ligase for cyclin D1, thereby preventing degradation of cyclin D1 in the cytoplasm and reimportation into the nucleus, where it exerts its oncogenic effects.21
In a similar vein, p16INK4a is an early genetic alteration, via promoter hypermethylation, point mutation, or allelic deletion, in Barrett’s esophagus and EAD, but interestingly, a late event in ESCC. Loss of heterozygosity of 9p21, the locus for both p16 and p15, has been demonstrated with high frequency in both dysplastic Barrett’s epithelium and Barrett’s adenocarcinoma (90% and more than 80% of cases, respectively).22,23 Promoter hypermethylation, which prevents tumor suppressor function by blocking transcription, has been documented and correlates with the degree of dysplasia in Barrett’s esophagus. It is present in up to 75% of specimens with high-grade dysplasia and is found in almost 50% of patients with adenocarcinoma of the esophagus.24 Point mutations of p16 in ESCC have been found and promoter hypermethylation has been noted in up to 50% of these tumors.25,26 Rb gene mutation is not found in either type of esophageal neoplasm, but allelic loss of 13q where the locus of the Rb gene resides is found in up to 50% of patients with Barrett’s adenocarcinoma and squamous cell carcinoma.18,27 This can correlate with diminished or loss of pRb protein in Barrett’s esophagus with dysplasia, EAD, and ESCC.28,29
p53 Tumor Suppressor Gene
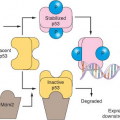
p53 is one of the most commonly mutated genes in human cancer.22,23,24 p53 (molecular weight approximately 53 kDa) is a tumor suppressor that interrupts the G1 phase to evaluate and permit repair of damaged DNA, which may arise from environmental exposure (e.g., irradiation, ultraviolet light) or cellular stress.30 In the face of irreparable damage, p53 induces apoptosis. The p53 transcription factor binds DNA to activate or suppress a large repertoire of target genes.31 p53 mutations induce loss of cell-cycle checkpoints and promote genomic instability. The majority of p53 mutations occur in the DNA-binding region, and more than 80% of them are missense mutations resulting in loss of wild type
p53 function.32 Wild type p53 has a short half-life and is difficult to detect by immunohistochemistry; mutation in p53 results in stabilization of the protein and allows for easier detection by immunohistochemistry.
p53 function.32 Wild type p53 has a short half-life and is difficult to detect by immunohistochemistry; mutation in p53 results in stabilization of the protein and allows for easier detection by immunohistochemistry.
Detection of mutated p53 protein by immunohistochemistry has been demonstrated with increasing frequency during histologic progression from Barrett’s esophagus (5%) through dysplasia (65% to 75%) to frank adenocarcinoma (up to 90%).33,34,35,36 Thus, p53 mutation or loss of heterozygosity appears early in Barrett’s esophagus and EAD. Both mutant p53 protein detected by immunohistochemistry and specific p53 gene mutations detected by genomic sequencing have been identified in 40% to 75% of patients with ESCC.37,38,39,40 The presence of a p53




Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
