Lung cancer tumorigenesis is a multistep process of transformation from normal bronchial epithelium to overt lung cancer. Various molecular events that result in gain or loss of function cause dysregulation of key genetic pathways involved in cellular proliferation, differentiation, apoptosis, migration, invasion, and other processes characteristic of the malignant phenotype. Mutations, including single nucleotide substitution or deletion, and translocation, deletion, or amplification of larger portions of genetic material may result from environmental factors, inherited susceptibility, or random events. Many genes are involved in tumorigenesis of both small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC) (Table 15.1, Fig. 15.1), but there are also unique genetic aberrations associated with each tumor type. Following the development of overt cancer, continued accumulation of genetic abnormalities influences the processes of invasion, metastases, and resistance to cancer therapy. Identification of the nature and frequency of these molecular abnormalities is necessary to determine their clinical implications (e.g., associations with smoking, histological type, stage, survival, response to therapy) and define their clinical utility for prevention and early diagnosis of lung cancer, as well as for the development of therapeutic targets.
SUSCEPTIBILITY TO LUNG CANCER: GENETIC SUSCEPTIBILITY AND CARCINOGENS IN TOBACCO SMOKE
Tobacco use is the most important environmental factor associated with the development of lung cancer. Approximately 85% of lung cancer occurs in current or former smokers, which corresponds to a greater than tenfold increase in risk of lung cancer compared to never-smokers. Cigarette smoke contains more than 60 known carcinogens, 20 of which have been convincingly shown to cause lung tumors in laboratory animals or humans.1 Of these, polycyclic aromatic hydrocarbons, such as benzo(a)pyrene, tobacco-specific nitrosamines, such as 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), and aromatic amines, such as 4-aminobiphenyl, appear to have an important role in cancer causation. Nitrosamines such as NNK induce lung tumors, primarily adenomas and adenocarcinomas, in mice independent of the route of administration. Among the polycyclic aromatic hydrocarbons, benzo(a)pyrene is the most extensively tested and the first to be detected in tobacco smoke. Its role in cancer tumorigenesis is well described, and its diol epoxide metabolite has been implicated as the cause of mutation in the TP53 gene.2 One of the carcinogenic effects of tobacco smoke in the lung is the formation of DNA adducts, leading to errors in DNA replication and resulting mutations. DNA adducts have been identified in the bronchial tissue of patients with lung cancer. In current smokers, adduct levels correlate with the amount of tobacco smoke exposure.3,4 Smoking cessation for at least 5 years results in adduct levels similar to nonsmokers.4 In addition, in former smokers, age at smoking initiation has been inversely associated with levels of DNA adducts, suggesting that prevention of smoking in adolescence is of utmost importance in decreasing lung cancer risks.
Although tobacco use can account for the majority of lung cancers, most chronic smokers still do not develop lung cancer. Differences in inherent susceptibility may be related to variations in carcinogen metabolizing enzymes, DNA repair mechanisms, chromosome fragility, and other homeostatic mechanisms. Among genes for carcinogen-metabolizing enzymes, polymorphisms in the cytochrome P-450 genes CYP1A1, CYP2D6, and CYP2E1 and in mu-class glutathione S-transferase (GSTM1) have received the most attention. Although studies have suggested that there may be a modest association of GSTM1 null polymorphism with lung cancer, knowledge of the state of single candidate genes may not be adequate to predict lung cancer risk due to the complexity of carcinogen metabolism, gene-gene and gene-environment interactions, and the relatively small effect of an individual gene. In addition to inherent susceptibility to the carcinogenic effects of tobacco smoke, large genome-wide association studies have identified lung cancer susceptibility loci at 15q25, 5p15, and 6p21.5,6 In particular, polymorphisms in and around nicotinic cholinergic receptors at chromosome 15q25 appear to correlate with messenger RNA (mRNA) and protein expression of these receptors as well as functional changes in the calcium ion channel of the A5 nicotine receptor; these differences confer susceptibility to smoking behaviors.7
TABLE 15.1 MOST FREQUENTLY ACQUIRED MOLECULAR ABNORMALITIES IN LUNG CANCER
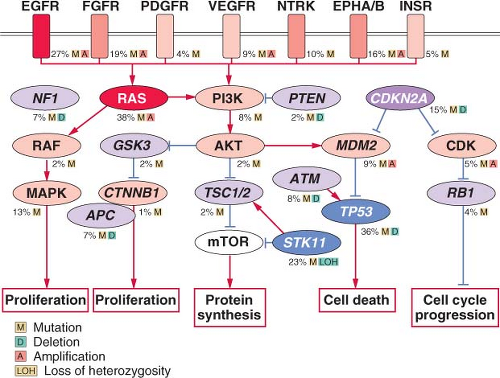
FIGURE 15.1 Significantly mutated pathways in lung adenocarcinomas. Genetic alterations in lung adenocarcinoma frequently occur in genes of the mitogen-activated protein kinase (MAPK) signaling, p53 signaling, Wnt signaling, cell cycle, and mammalian target of rapamycin (mTOR) pathways. Oncoproteins are indicated in pink to red and tumor suppressor proteins are shown in light to dark blue. The darkness of the colors is positively correlated to the percentage of tumors with genetic alterations. Frequency of genetic alterations for each of these pathway members in 188 tumors is indicated. (From Macmillan Publishers Ltd, ref. 113, copyright 2007, with permission.).
Researchers are optimistic that molecular epidemiology will help identify individuals at the highest risk of developing lung cancer. Such information, in addition to the smoking history, will be of great value in new lung cancer screening trials and in chemoprevention trials to identify persons at highest risk of developing lung cancer.
MOLECULAR CHANGES IN PRENEOPLASIA
Before lung cancer is clinically recognizable, a series of morphologically distinct changes (hyperplasia, metaplasia, dysplasia, and carcinoma in situ) are thought to occur. Whether one cell of origin leads to all histological variants is unclear. It is believed that dysplasia and carcinoma in situ represent true preneoplastic changes. These sequential changes found within squamous cell cancers that arise from central bronchi have long been recognized. Although the exact cell of origin for lung cancer is unknown, it is thought that type II epithelial cells have the capacity to give rise to lung adenocarcinomas, while cells of neuroendocrine origin are likely precursors of SCLC.
It is evident that preneoplastic cells contain several genetic abnormalities identical to some of the abnormalities found in overt lung cancer cells. For squamous cell cancers, immunohistochemical analysis has confirmed abnormal expression of protooncogenes (cyclin D1) and tumor suppressor genes (TSGs) (p53).8 Allelotyping of precisely microdissected, preneoplastic foci of cells shows that 3p allele loss is currently the earliest known change, suggesting that one or more 3p TSGs may act as gatekeepers for lung cancer pathogenesis.9 This is followed by 9p, 8p, and 17p allele loss and p53 mutation. Even histologically normal bronchial epithelium has been shown to have genetic losses. Similarly, atypical alveolar hyperplasia, the potential precursor lesion of adenocarcinomas, can harbor Kristen rat sarcoma viral oncogene homolog (KRAS) mutations and allele losses of 3p, 9p, and 17p.10 Other genetic alterations, such as inactivation of LKB1, whose germline mutations cause Peutz-Jeghers syndrome, have also been implicated in the development of adenocarcinoma. These observations are consistent with the multistep model of carcinogenesis and a field cancerization process, whereby the whole tissue region is repeatedly exposed to carcinogenic damage (tobacco smoke) and is at risk for development of multiple, separate foci of neoplasia.11
Although all types of lung cancers have associated molecular abnormalities in their normal and preneoplastic lung epithelium, SCLC patients in particular appear to have multiple genetic alterations occurring in their histologically normalappearing respiratory epithelium. Molecular changes have been found not only in the lungs of patients with lung cancer but also in the lungs of current and former smokers without lung cancer. These molecular alterations are thus important targets for use in the early detection of lung cancer and for use as surrogate biomarkers in following the efficacy of lung cancer chemoprevention. In this regard, it appears that the smoking-damaged respiratory epithelium has thousands of clonal patches, each containing clones of cells with 3p and other allele loss abnormalities.12 The challenge is to identify not only the prevalence and temporal sequence of molecular lesions in lung preneoplasia, but to determine which are rate limiting and indispensable and thus represent potential candidates for intermediate biomarker monitoring and therapeutic efforts.
GENETIC AND EPIGENETIC ALTERATIONS IN LUNG CANCERS
Genomic Instability and DNA Repair Genes
Similar to other epithelial tumors, lung cancer cells typically display chromosomal instability—both numeric abnormalities (aneuploidy) of chromosomes and structural cytogenetic abnormalities.13 Allele loss on chromosome 3p is thought to be the among the earliest genetic change occurring in both NSCLC and SCLC.14 In addition, nonreciprocal translocations and recurrent losses involving 1p, 4p, 4q, 5q, 6p 6q, 8p, 9p, 11p, 13q, 17p, 18q, and 22q may occur, representing changes in known and potential tumor suppressor genes.15 Polysomies or regions of gene amplifications also occur and often involve protooncogenes such as epidermal growth factor receptor (EGFR) and myelocytomatosis viral oncogene homolog (MYC).16,17 Simple reciprocal translocations are uncommonly observed in lung cancer, although translocations that give rise to BRD4-NUT,18 CRCT1-MAML2,19 SLC34A2-ROS,20 and EML4-ALK21 fusion proteins have been reported, and for the case of EML4-ALK this has been shown to drive the development and proliferation of tumors. Alterations in microsatellite polymorphic repeat sequences are found in 35% of SCLC and 22% of NSCLC.22 The underlying mechanism for this chromosomal instability has not yet been discovered.
The most powerful tumor surveillance mechanism is involved in DNA damage response and repair of errors in DNA replication.23 The DNA glycosylase 8-Oxo guanine (OGG1) specifically excises the oxidatively damaged mutagenic base 8-hydroxyguanine, which causes G:C→T:A transversions frequently found in lung cancer. Lung adenocarcinoma spontaneously develops in Ogg1 knockout mice and 8-hydroxyguanine accumulates in their genomes.24,25 Individuals with low OGG activity have a greatly increased risk of developing lung cancer. Polymorphisms in other DNA repair genes ERCC1, XRCC1, ERCC5/XPG, and MGMT/AGT have been correlated with reduction of polyaromatic hydrocarbon DNA adduct formation as well as with lower lung cancer risks in case-control studies. High expression of excision repair cross-complementation gene-1 (ERCC1) is associated with decreased response to platinum-based chemotherapy, but in contrast, overexpression of ERCC1 correlates with better overall prognosis in NSCLC,26 reflecting improved repair of lethal DNA damage by platinum, on the one hand, and greater DNA stability with less aggressive disease course, on the other. Similarly, ribonucleotide reductase M1 (RRM1) overexpression correlates with better de novo prognosis but resistance to gemcitabine. Assays of both ERCC1 and RRM1 are commercially available and are marketed as tools to guide cytotoxic therapy in NSCLC; treatment based on these assays has not yet been prospectively validated, but may prove important.
PROTOONCOGENES, GROWTH FACTOR SIGNALING, AND GROWTH FACTOR TARGETED THERAPIES
EGFR and KRAS are the two most commonly mutated protooncogenes in lung adenocarcinomas; these mutations appear to be mutually exclusive. EGFR is a transmembrane tyrosine kinase, which, when activated by binding with one of its ligands, members of the EGF family, stimulates cell proliferation. When mutated, EGFR tyrosine kinase is constitutively activated, resulting in uncontrolled proliferation, invasion, and metastasis. Coexpression of EGFRs and their ligands, especially transforming growth factor-α, by lung cancer cells indicates the presence of an autocrine (self-stimulatory) growth factor loop. Activating EGFR mutations are observed in approximately 10% of North American and European populations and 30% to 50% of Asian populations27,28,29,30,31,32 and are significantly more common in never-smokers (100 or less cigarettes per lifetime) or light former smokers (quit 1 year or more ago and less than ten-pack per year smoking history). The leucine to arginine substitution at position 858 (L858R) in exon 21 and short in-frame deletions in exon 19 are the most common mutations seen in adenocarcinomas of the lung. These mutations result in prolonged activation of the receptor and downstream signaling through phosphorylated Akt, in the absence of ligand stimulation of the extracellular domain. EGFR mutations are both prognostic for response rate to chemotherapy and survival irrespective of therapy and are predictive of response to specific inhibitors of the EGFR tyrosine kinase—gefitinib and erlotinib.31,33,34EGFR mutations are found almost exclusively in adenocarcinomas and occur much more frequently in tumors from never-smokers, women, and in Asian populations, explaining the increased clinical response to gefitinib noted in these subpopulations before the association with EGFR mutation was discovered.31,33,34 A recent review by Rosell et al.32 suggested patients with adenocarcinoma who are never or light remote smokers, especially women and Asians, should be screened for the presence of an EGFR mutation. The IPASS (Iressa Pan-Asia Study) trial demonstrated an improved outcome with up-front treatment with gefitinib compared with chemotherapy for patients with EGFR mutation, but on the other hand, much worse outcome compared with chemotherapy in patients with wild type EGFR.35,36 Previous suggestions that EGFR gene amplification by detected by fluorescence in situ
Only gold members can continue reading. Log In or Register to continue