Eric C. Johannsen, Kenneth M. Kaye
Epstein-Barr Virus (Infectious Mononucleosis, Epstein-Barr Virus–Associated Malignant Diseases, and Other Diseases)
Epstein-Barr virus (EBV) is a ubiquitous human herpesvirus. Infection with EBV is common, worldwide in distribution, and largely subclinical in early childhood. EBV has been established as the causative agent of heterophile-positive infectious mononucleosis, which occurs most frequently in late adolescence or early adulthood. In addition, EBV is causally associated with the development of malignant diseases, including Burkitt’s lymphoma, lymphoproliferative disease, Hodgkin’s lymphoma, primary central nervous system (CNS) lymphomas in acquired immunodeficiency syndrome (AIDS), and nasopharyngeal carcinoma based on seroepidemiologic data and the detection of EBV genomes in these tumors. Some epidemiologic studies describe an association between EBV and autoimmune diseases, particularly multiple sclerosis; however, a causal relationship is not established.
History
Historical accounts of infectious mononucleosis often attribute the initial description of the disease to Filatov or Pfeiffer, who nearly simultaneously at the end of the 19th century described an illness characterized by malaise, fever, hepatosplenomegaly, lymphadenopathy, and abdominal discomfort.1,2 This illness came to be known as Drusenfieber (glandular fever) and occurred in family outbreaks. However, without specific techniques with which to establish the diagnosis, the concept of Drusenfieber as a clinical entity fell into disrepute. Between 1910 and 1920, a number of observers reported cases of apparent spontaneous remission of leukemia, with a clinical course that is consistent with the spontaneous resolution of infectious mononucleosis.3,4 The establishment of infectious mononucleosis as a clinical entity is credited to Sprunt and Evans,5 who in 1920 described six cases of fever, lymphadenopathy, and prostration that occurred in previously healthy young adults. The authors pointed out the mononuclear lymphocytosis that developed in each of the patients and contrasted the pathologic appearance of these lymphocytes with the uniform lymphocyte morphology observed in children with other infections. Two years later, Downey and McKinlay6 described additional cases of infectious mononucleosis and provided a more detailed morphologic description of the atypical lymphocyte. The recognition of atypical lymphocytosis as a hematologic marker for the disease led to more accurate descriptions of the clinical manifestations of this illness.
A major advance occurred in 1932, when Paul and Bunnell,7 investigating immunologic mechanisms in serum sickness, unexpectedly encountered high titers of spontaneously occurring sheep red blood cell agglutinins (heterophile antibodies) in the sera of patients with infectious mononucleosis.
During the 1940s and 1950s, substantial efforts were made to detect a causative agent for infectious mononucleosis. Attempts to culture etiologically related bacteria and viruses from patients with infectious mononucleosis proved unsuccessful. The disease could not be transmitted to animals. Interpretation of experimental attempts to transmit the disease to humans was hindered by the failure to appreciate the widespread occurrence of asymptomatic infection in preadolescents and the absence of a serologic marker of immunity.8–10
The identification of EBV followed the description by Burkitt11 in 1958 of an unusual lymphoma with a predilection for the head and neck. The geographic distribution of this tumor paralleled that of certain mosquito-borne diseases in Africa, and a search for an etiologically related arbovirus was undertaken. Epstein and associates12 in 1964 described the presence of particles that resembled herpesviruses in tissue cultures of biopsy specimens from patients with Burkitt’s lymphoma. An indirect immunofluorescent assay for detecting anti-EBV antibodies was developed by Werner and Gertrude Henle,13 and high titers were detected in patients with Burkitt’s lymphoma. Additional studies revealed that 90% of American adults had demonstrable EBV antibodies as well.13 The development of infectious mononucleosis in a technician in the Henles’ laboratory on whom sequentially obtained sera were analyzed for EBV antibody suggested that acute EBV infection may be associated with this illness.14 Large-scale epidemiologic studies15–18 showed that heterophile-positive infectious mononucleosis occurred in patients without preexisting EBV antibody, and, conversely, heterophile-positive infectious mononucleosis was always accompanied by acquisition of EBV antibodies. These epidemiologic studies indicated that subclinical EBV infection also occurred. With specific antibody tests for EBV, it became apparent that 10% to 20% of the cases of mononucleosis, of which most were heterophile negative, were caused by other agents, the most frequent of which was cytomegalovirus (CMV). This chapter deals primarily with EBV-induced infectious mononucleosis.
Description of Epstein-Barr Virus
Physical Properties
EBV, or human herpesvirus 4, is a gamma-1 herpesvirus. Like the other members of the Herpesviridae family, EBV has a double-stranded DNA genome encased in an icosahedral protein nucleocapsid surrounded by a lipid envelope embedded with viral glycoproteins. Herpes-viruses also have an amorphous protein layer, the tegument, which lies between the capsid and envelope. The B95-8 laboratory strain of EBV, the first herpesvirus genome sequenced, was found to have a 12-kilobase (kb) deletion and the wild-type EBV genome, which is approximately 172 kb in size, encodes about 90 proteins and 25 microRNAs.19,20
Life Cycle
Primary infection with EBV results from exposure to the oral secretions of seropositive individuals through kissing, sharing of food, or other intimate contact. The long-accepted concept that EBV infection spreads to B lymphocytes after initial productive (lytic) infection of oral epithelial cells21,22 has been challenged. Tonsillar biopsies from patients with primary EBV infection did not reveal any infected epithelial cells, but infected lymphocytes were readily seen.23,24 EBV undoubtedly has clinically significant tropism for epithelial cells, as is seen in nasopharyngeal carcinoma and oral hairy leukoplakia. It remains possible that significant infection of oral epithelial cells occurs in nontonsillar sites or that an initial round of lytic replication precedes spread to the B-cell compartment and the onset of symptoms.25,26 Infected B lymphocytes incite an intense cytotoxic T-cell response, and these T cells constitute the atypical lymphocytosis characteristic of primary EBV infection.27,28 In healthy individuals, most infected B lymphocytes are cleared through immune surveillance, but between one and 50 B cells per million remain quiescently infected and serve as the reservoir for lifelong infection of the individual.29,30 Thus, EBV shares the properties of lifelong latency and persistence with other members of the herpesvirus family. In contrast to that of alpha herpes viruses (herpes simplex virus and varicella-zoster virus), shedding of infectious EBV particles into the saliva from periodic reactivation of latently infected cells is entirely asymptomatic. This shedding occurs in otherwise healthy persons but is more frequent in immunosuppressed hosts (Table 141-1).
TABLE 141-1
Frequency of EBV Shedding
| POPULATION DESCRIPTION | OROPHARYNGEAL SHEDDING RATE (RANGE) | REFERENCE |
| EBV-seronegative individuals | 0 | 89 |
| Seropositive healthy adults | 12%-25% | 85–87, 89–91 |
| Solid tumor patients | 27% | 86, 87 |
| HIV-1–infected individuals | 50% | 88 |
| Renal transplant recipients | 56%-70% | 85, 87 |
| Infectious mononucleosis patients | 50%-100% | 89–91, 348 |
| Critically ill leukemia or lymphoma patients | 74%-92% | 86, 87 |
EBV, Epstein-Barr virus; HIV, human immunodeficiency virus.
The host range of the virus is limited. In vitro cultivation of the virus has been described primarily in B lymphocytes and also in nasopharyngeal epithelial cells of humans and certain nonhuman primates.31 EBV binds to its receptor, the CD21 molecule, through an interaction with its major envelope glycoprotein, gp350. CD21 or complement receptor 2 (CR2) transduces signals important for B lymphocyte proliferation and can also be expressed by follicular dendritic cells and nasopharyngeal epithelial cells.32–38 Another EBV glycoprotein, gp42, binds major histocompatibility complex (MHC) class II molecules, which serve as co-receptors for infection of B cells.39–43 Gp42 also promotes B-cell infection by forming a heterotrimeric complex with the gH/gL EBV glycoproteins, masking a motif on gH that is important for infection of epithelial cells.44 EBV virions released from infected B lymphocytes contain lower levels of gp42 and infect epithelial cells more efficiently than virions derived from epithelial cells. This reciprocal tropism is proposed to enhance EBV shuttling between B lymphocytes and the oral epithelium.45
Latent Infection and Growth Transformation
After infection with EBV, B lymphocytes enter the cell cycle and proliferate continuously in a process termed transformation or immortalization; these cells can be propagated in vitro indefinitely.46 This ability of EBV to convert peripheral blood B cells into immortalized lymphoblastoid cell lines (LCLs) is widely used in genomic studies as a means of preserving DNA samples from volunteer donors for future use.47 In vivo EBV-driven B-cell proliferation is observed during infectious mononucleosis, in which it probably serves to expand rapidly the pool of infected B lymphocytes. These B lymphocytes are usually rapidly cleared from the circulation.48–51 However, in the absence of an intact immune response, EBV infection can result in life-threatening lymphoproliferative disease (LPD).52,53 The growth-transforming properties of EBV can act in concert with genetic and environmental cofactors to cause malignant diseases in immunocompetent hosts as well.54,55
EBV infection of B lymphocytes is characterized by a state of viral latency, in which the genome circularizes in the nucleus and is replicated as an episome in concert with host chromosomes by cell enzymes. The infection is latent in the sense that viral particles are not being produced, but it is anything but quiescent. Limited viral gene expression persists, and these genes exert effects on the infected cell. In vitro latent infection of B lymphocytes with EBV is characterized by the expression of latent infection membrane proteins 1 and 2 (LMP1 and LMP2), six EBV nuclear antigens (EBNAs), and two small, nuclear, noncoding RNAs (EBV-encoded RNAs [EBERs]) that are transcribed by RNA polymerase III (Table 141-2).56 Additional EBV transcripts have been detected in latent infection and are termed complementary strand transcripts (CSTs) or BamH1 A rightward transcripts (BARTs). Translation of these transcripts into proteins has not been shown, but they appear to serve as precursors for two of three clusters of EBV-encoded microRNAs (miRNAs) whose role in EBV biology remains unclear.57,58 Recombinant reverse genetic analysis has determined that only LMP1, EBNA1, EBNA2, EBNA3A, EBNA3C, and EBNALP are critical for B-cell growth transformation.56 The mechanisms by which these EBV gene products promote B-lymphocyte growth have been the subject of intense investigation.
TABLE 141-2
Patterns of EBV Latent Gene Expression
| EBV GENE PRODUCT | FUNCTION | EBV-ASSOCIATED MALIGNANT DISEASES | |||||||
| Acute Infection | Healthy Carrier | Latency III | Latency II | Latency I | |||||
| IM | PBB | LPD | PCNSL | HL | NPC | GC | BL | ||
| EBNA1 | EBV genome maintenance | + | ? | + | + | + | + | + | + |
| EBNA2 | Activates expression of EBV/host genes | + | − | + | + | − | − | − | − |
| EBNA3s* | Represses p16/INK4A tumor suppressor expression | + | − | + | + | − | − | − | − |
| EBNALP | Coactivates with EBNA2 | + | − | + | + | − | − | − | − |
| LMP1 | Mimics CD40 signaling | + | − | + | + | + | + | − | − |
| LMP2 | Mimics BCR signaling | + | + | + | + | + | + | ± | − |
| miRNAs | Block expression of host RNAs | + | ? | + | + | + | + | + | + |
| EBERs | Noncoding, highly expressed RNAs | + | + | + | + | + | + | + | + |
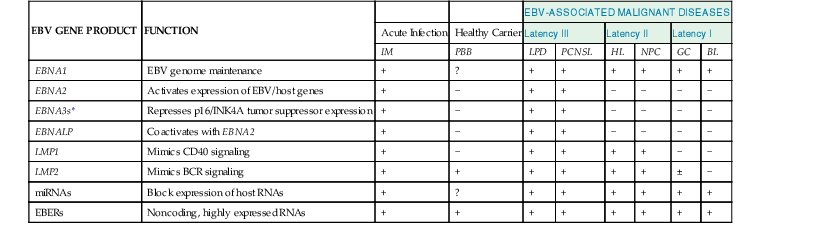
* Includes EBNA3A, EBNA3B, and EBNA3C.
BCR, B-cell receptor; BL, Burkitt’s lymphoma; EBERs, EBV-encoded RNAs; EBV, Epstein-Barr virus; GC, gastric cancer; HL, Hodgkin’s lymphoma; IM, infectious mononucleosis; LPD, lymphoproliferative disease; NPC, nasopharyngeal carcinoma; PBB, peripheral blood B cell; PCNSL, primary central nervous system lymphoma.
Note: Because EBV-positive gastric cancers sometimes express LMP2 and, rarely, LMP1, these tumors have been classified as latency I or II by different experts.
After the virus gains entry to susceptible B lymphocytes, EBNA2 and EBNALP are the first proteins expressed. EBNA2 is an acidic transactivator that acts as the major switch to turn on latent virus gene expression and several B-cell gene products (including c-myc, c-fgr, CD21, and CD23). It has no intrinsic sequence-specific DNA binding capacity but rather is targeted to promoters by binding to a host DNA binding protein RBP-Jκ (also called CBF1 or CSL), a downstream component of the Notch signaling pathway.59,60 By an incompletely understood mechanism, EBNALP cooperates with EBNA2 to activate expression of the remaining nuclear proteins and LMP1 and LMP2.61 LMP1 is the major EBV-encoded oncogene, and its expression in transgenic mice results in B-cell lymphomas.62,63 It constitutively activates signaling pathways that mimic the growth and survival signals given to B cells by CD4+ T lymphocytes through the CD40 surface glycoprotein. LMP1 sends this signal through its cytoplasmic tail, which binds a set of second messenger proteins similar but not identical to those used by CD40.64,65 Unlike CD40, LMP1 does not require the presence of ligand to form patches in the cell membrane but self-associates constitutively, approximating its cytoplasmic tails to activate signaling.66 This results in the activation of nuclear factor κB (NF-κB); c-jun; upregulation of adhesion molecules (intercellular adhesion molecule 1, LFA-1, and LFA-3); cytokine production; B-cell proliferation; and induction of an antiapoptotic state.48,55 A second EBV latent membrane protein, LMP2, mimics another signal necessary for B-cell survival.67 By interacting with signaling molecules of the B-cell receptor (BCR), LMP2 mimics BCR engagement by constitutive patching in the membrane in a manner analogous to LMP1. LMP2 probably also interferes with normal signaling through the BCR by antigenic stimulation to inhibit activation of lytic viral replication (discussed subsequently). Interestingly, LMP2 is not necessary for EBV-mediated outgrowth of B cells in vitro but is probably a critical component of the viral strategy in vivo. The nuclear protein EBNA1 acts to promote the replication of the viral genome by the host machinery when the virus is in the latent, episomal state and to ensure proper segregation of the EBV genome to both daughter cells. The EBNA3 proteins also interact with the RBP-Jκ DNA binding protein and promote B lymphocyte growth and survival by silencing expression of the p16INK4 and p14ARF tumor suppressor gene products.68,69 The function of the highly expressed, noncoding EBV RNAs (EBERs) is incompletely understood.
EBV-associated malignant diseases are exclusively associated with latent infection and latent gene expression. Three general patterns of expression of EBV-encoded proteins have been observed in association with latency (see Table 141-2).48,49 Expression of all latent genes is seen in LPD in immunosuppressed hosts, in primary CNS lymphoma of patients with AIDS, and during primary EBV infection (infectious mononucleosis), and this program of gene expression is often referred to as latency III.54 EBV-associated nasopharyngeal carcinomas, Hodgkin’s lymphoma, and T-cell lymphomas exhibit a more restricted pattern of EBV gene expression (latency II) that includes LMP1, LMP2, EBNA1, and the EBERs and EBNA1.70,71,72 In Burkitt’s lymphoma (latency I) only EBERs and EBNA1 are expressed.54 The more restricted patterns of latent gene expression in some tumors are probably in part the result of the intense immune response against viral proteins.
Lytic Infection
Latent infection can be activated to lytic infection by stimulation of host B cells by certain chemicals, calcium ionophores, or antibodies to surface immunoglobulin.73 The physiologic signals that reactivate EBV lytic replication are unknown, but signaling through the B-cell receptor after antigenic stimulation is a possible scenario. After this inciting event, two EBV-encoded transcriptional activators are expressed: BZLF1 and BRLF1. Expression of these immediate early genes leads to a cascade of events that culminate in the production of early EBV gene (early antigen [EA]) products responsible for viral replication (e.g., thymidine kinase, DNA polymerase) and late (structural) genes of the virus including viral capsid antigens (VCAs).74 Lytic infection produces EBV virions and can cause host cell death.
Epidemiology
Serum Antibody Prevalence
Antibodies to EBV have been found in all population groups studied, and most studies have shown no predilection for either gender. Antibodies are acquired earlier in life in developing countries than in industrialized countries, but by adulthood, 90% to 95% of most populations have demonstrable EBV antibodies.75,76 In the United States and in Great Britain, EBV seroconversion occurs before the age of 5 years in about 50% of the population.76–78 A second wave of seroconversion occurs midway through the second decade of life. EBV seroconversion may occur at a younger average age in the southern United States than in other areas of that country.79 Lower socioeconomic groups have a higher EBV antibody prevalence than more affluent age-matched control groups.
Two strains of EBV have been defined on the basis of viral gene sequences expressed during latency and their ability to transform B lymphocytes.73 The strains (type 1 [A] or 2 [B]) are not distinguishable serologically, but they express unique epitopes that are identified by cytotoxic T lymphocytes (CTLs). Although the initial thought was that specific geographic distributions existed for these two strains of EBV, it is now clear that both are widely distributed and that individuals can be coinfected with both strains.
Incidence of Infection
Clinically apparent infectious mononucleosis is more common in populations in which primary EBV exposure is delayed until after the first decade of life. The disease is diagnosed most frequently among adolescents of higher socioeconomic groups in industrialized countries.80 The incidence of infectious mononucleosis in a large epidemiologic study in the United States was 45.2 cases per 100,000 per year and was highest in the 15- to 24-year-old age group.81 The incidence was the same for women as for men, but the peak age-specific incidence occurred 2 years earlier in women. Infectious mononucleosis is 30 times more frequent in whites than in blacks. The infrequency of infectious mononucleosis among blacks, noted as early as 1940, is probably a reflection of earlier primary EBV infection and the higher frequency of subclinical infections in children.82–84 No clear seasonal incidence has been noted.
Methods of Spread
The virus persists in the B-cell compartment for the life of the infected host and can be cultured from throat washings from 10% to 20% of healthy adults, from 50% of kidney transplant recipients, and from greater proportions of those critically ill with leukemia or lymphoma (see Table 141-1).85–87 Approximately 50% of men with human immunodeficiency virus type-1 (HIV-1) infection who have sex with men shed EBV in oropharyngeal secretions.88 Low titers of EBV are present in throat washings of persons with infectious mononucleosis.89–91 Susceptible roommates of students with infectious mononucleosis or with inapparent EBV infection have EBV seroconversion no more frequently than the general susceptible college population.17,79 Only 6% of those with infectious mononucleosis cite previous contact with another case of infectious mononucleosis.81 EBV DNA or protein, or both, have also been identified in parotid duct and uterine cervical epithelia, although the implications of this distribution are unclear with respect to viral transmission.92,93
EBV, like other herpesviruses, is relatively labile in the laboratory, and the virus has not been recovered from environmental sources, including fomites. These data suggest that EBV is a widespread agent that is not particularly contagious and that most cases of infectious mononucleosis are probably contracted by intimate contact between susceptible individuals and asymptomatic shedders of EBV. Among young adults, spread of the virus may be facilitated by the transfer of saliva with kissing.94,95 EBV infection has been linked to sexual intercourse; however, other studies have found that kissing with coitus conferred no additional risk of EBV infection compared with kissing without coitus.96,97 Serologic evidence suggests that the virus may also be spread among susceptible individuals within families.98,99 EBV has also been spread via blood transfusion and after open heart surgery as the postpump perfusion syndrome.100 Most postpump perfusion infectious mononucleosis is, however, heterophile negative and attributable to CMV.
Although several apparent epidemics of infectious mononucleosis have been described, these reports have not been substantiated with EBV serologic data and have lacked rigorous epidemiologic, clinical, or laboratory support. Some of these have resulted from errors in the performance of Monospot tests.101 On the basis of the previously discussed information, true epidemics of infectious mononucleosis are unlikely to occur.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


