SOME EPIGENETIC MODIFICATIONS AND THEIR ROLES IN GENE ACTIVITIES
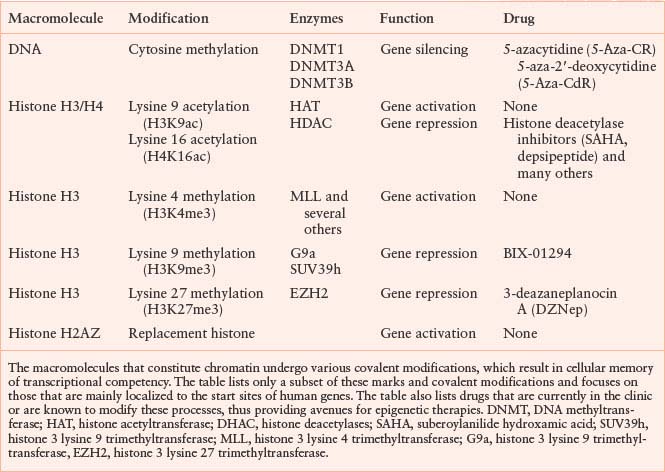
Figure 3.1 depicts the positions of a small subset of the possible modifications on the histone H3 protein in the context of nucleosomes. Although there are other modifications such as phosphorylation, ubiquitination, and sumolation of this and other histones, the discussion here is restricted to methylation and acetylation, since their function and potential for drug development is currently best understood. The various modifications can be interpreted by other proteins (not shown) sometimes called “readers,” which modify local chromatin structure to either stimulate or repress gene expression. Still other proteins (also not shown), such as histone deacetylases or histone demethylases, can remove the modifications in response to cellular and environmental signals, resulting in a dynamic state.
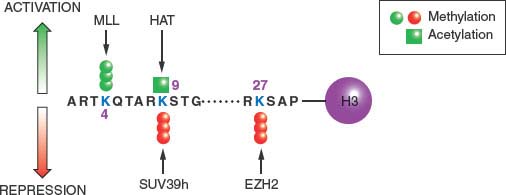
FIGURE 3.1 Covalent modifications of histones can regulate gene activity. The location of activating and repressive marks on histone H3 are shown as an example. These covalent modifications, including trimethylation of lysine 4 (K4me3) and acetylation of lysine 9 (K9ac), are highly localized in start sites of genes and associated with active gene transcription. Conversely, methylation of lysine 9 (K9me3) or lysine 27 (K27me3) is associated with gene inactivity. It is the balance between these marks that define the transcriptional competence of a given gene. Unlike the two activating marks shown (green), the two repressive marks (red) tend to be more widely distributed on chromatin and potential drug targets. The figure is not intended to be comprehensive, and many additional modifications on histone H3 and other histones are known to participate in the structure of the epigenome. MLL, histone 3 lysine 4 trimethyltransferase; HAT, histone acetyltransferase; SUV39h, histone 3 lysine 9 trimethyltransferase; EZH2, histone 3 lysine 27 trimethyltransferase.
The positioning of the modifications relative to the TSS of genes is also critical for their function: Figure 3.2. shows the start site of a hypothetical gene with a CpG island in its promoter, which is normally free of DNA cytosine methylation. Active genes attach the activating H3K4me3 and H3K9ac modifications to the nucleosomes flanking the TSS. These may serve as “beacons,” allowing the transcriptional apparatus to “find” the start site and begin producing mRNA.
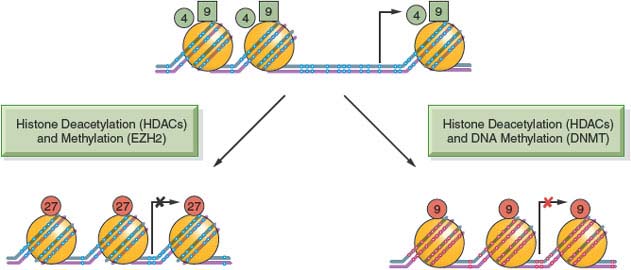
FIGURE 3.2 DNA methylation, histone modifications, and nucleosome occupancy define active and silenced states. The figure depicts a hypothetical CpG island that is active in gene expression and contains unmethylated CpG sites (white dots) within the CpG island and the presence of active histone H3 marks, including lysine 4 methylation (H3K4me3) (green circles) and lysine 9 acetylation (H3K9Ac) (green squares). Genes repressed by histone deacetylation and methylation by EZH2 show the insertion of a nucleosome (large orange circles) into the transcriptional start site and the application of lysine 27 trimethylation (H3K27me3) (red circles). This state is commonly observed in embryonic stem cells and holds genes in a poised silent state so that they can be called upon later for expression during embryogenesis and cell differentiation. The CpG island can also become silenced in a more permanent manner by histone deacetylation and DNA methylation, resulting in the presence of 5-methylcytosine (red dots) near the transcriptional start site. The histone mark associated with this state is often (but not exclusively) lysine 9 methylation (red circles). Genes silenced by this mechanism tend to be permanently silenced, and examples include genes on the inactive X-chromosome in mammalian cells.
The silencing of the gene can be brought about in several ways, such as the removal of the activating H3K4me3 and H3K9ac modifications and application of the H3K27me3 mark (Fig. 3.2). Insertion of a nucleosome into the nucleosome-free region characteristic for CpG island–containing genes may also facilitate the silencing process. Genes silenced through DNA cytosine methylation also have nucleosomes at the start site of the gene but include the H3K9me3 modification rather than the H3K27me3 modification as a distinguishing feature.4 This more permanent mitotically heritable silencing process is used to ensure the long-term silencing of X-chromosome–linked genes and other important genes such as imprinted genes throughout the life of a human. These silencing mechanisms are all essential for mammalian development and maintenance of normal physiologic functions and can become pathologically altered in cancer and precancerous conditions, leading to widespread mitotically heritable aberrations in gene expression that characterize the cancer state.
Epigenetic processes such as those discussed above not only play important roles during development, but also are involved in maintaining tissue-specific patterns of gene expression in differentiated cells. In particular, DNA methylation patterns vary in different cell types, particularly at the transcription start sites of genes that do not contain CpG islands. Although the exact relations between these methylation patterns and control of gene expression have not been completely worked out, they probably assist in mitotic maintenance of differentiated states.
EPIGENOMIC CHANGES IN CANCER
DNA methylation patterns and histone modifications are essential for physiologic processes yet their dysregulation contributes to the cancer process. Epigenomic changes tend to be self-reinforcing and progressive and arise in normal cellular processes such as aging, which is one of the strongest risk factors for cancer. Several elements such as key genes that control the integrity of the genome (e.g., tumor suppressor genes, the adhesiveness of cells, the regulation of cell division, and the execution of apoptotic pathways) are all subject to inappropriate epigenetic silencing in cancer cells.
The field of cancer epigenetics started more than three decades ago with the observation that DNA methylation levels were profoundly altered in cancer cells relative to their normal counterparts.5 Subsequently, it became clear that the overall hypomethylation of the genome observed in cancer relative to normal cells was accompanied by focal hypermethylation near the TSS of key regulatory genes such as tumor suppressor genes.6 This focal hypermethylation of the CpG island regions at the TSS of the genes is associated with mitotically heritable silencing. Because of the inherent ability of DNA methylation patterns to be copied over a protracted time period, it was soon realized that these methylation changes could result in a molecular pathway that satisfies Knudson’s hypothesis for the inactivation of tumor suppressor genes. Knudson hypothesized that at least two hits were required for the inactivation of tumor suppressor genes in familial cancers such as retinoblastoma (RB).7 He proposed that mutations in the coding regions of genes such as the RB gene, followed by a loss of the wild-type copy through various pathways, could give rise to the cancer phenotype. However, we now know that inappropriate methylation of the TSS of a gene can also result in its mitotically heritable silencing and give rise to the cancer phenotype.6 This hypothesis has been confirmed in several human cancers, including gastric cancers in Pacific Islanders. In these cases, families bearing a germline mutation in one allele of the E-cadherin gene show a high propensity for developing gastric cancer at later stages in their lives.8 In many cases, the wild-type allele of the gene had become inappropriately silenced by aberrant DNA methylation, therefore leading to the initiation of the carcinogenic process in the stomach epithelium.
Widespread alterations in DNA methylation patterns in specific genes known to participate in human carcinogenesis have been identified in numerous studies. In addition, methylation-induced dysregulation of genomic imprinting is also found in cancer. Loss of imprinting of IGF2 and other imprinted genes have been associated with various childhood and adult cancers. The development of new high-throughput sequencing technologies has resulted in the discovery of even more loci that undergo epigenetic changes, some of which are relevant to the cancer phenotype. This has allowed identification of specific epigenetic cancer signatures such as the CpG island methylator phenotype (CIMP) in sporadic colorectal cancer.9 Many of these changes occur as a function of aging, which may explain the increased risk of various cancers in older individuals.10 Epidemiologic studies are beginning to reveal the influence of environmental factors on the epigenome11 and have identified epigenetic alterations as contributing mechanisms, linking lifestyle factors and cancer incidence and prognosis.12,13 Future studies with improved design, including larger sample sizes, defined study populations, and control for confounding variables, will allow identification of the key loci affected by aberrant methylation in a substantial proportion of individuals affected by specific cancer types.14
Substantial alterations in DNA methylation pattern have also been found in the apparently normal tissues of individuals with infections. Perhaps the best characterized example is the gastric epithelium of patients infected by Helicobacter pylori. The infection leads to hypermethylation of the TSS of specific genes that are lost when the infection is cured by suitable antibiotic intervention.15 Since the antibiotics do not cause demethylation directly, the loss of hypermethylation in these individuals is probably due to the loss of chronic inflammation. Since infections such as H. pylori, human papilloma virus, and other viral infections are causal players in carcinogenesis, these changes suggest that epigenetic alterations may play pivotal roles in the formation of cancers in infected individuals.
More recently with the development of chromatin immunoprecipitation (ChIP), coupled with high-throughput sequencing (ChIP-Seq), it has been realized that changes in DNA methylation are only part of the epigenomic alterations in cancer cells. Widespread alterations in the chromatin of cancer cells include genes that, although not silenced by DNA methylation, are repressed by histone modifications such as the application of aberrant patterns of H3K27me3 by the enzyme EZH2.16 Although these changes may not be as mitotically stable as those induced by DNA hypermethylation, they nevertheless represent important therapeutic targets. Likewise, genes silenced by histone acetylation can be reactivated by treatment with histone deacetylase drugs. Thus, the field of cancer epigenetics is moving into a more holistic appreciation of epigenome-wide changes that occur in human cancer and an understanding of how these dysregulations contribute to cancer pathology.
THE TIMING OF EPIGENETIC ALTERATIONS
Aberrant epigenetic gene silencing probably occurs at a very early stage in neoplastic development. Epimutations may therefore be initiators in carcinogenesis, allowing for the early clonal expansion of cells subsequently at risk for additional alterations. The abnormal silencing of epigenetic “gatekeepers” might be induced by risk factors such as aging and inflammation.17 Such gatekeepers normally restrict the division potential of stem cells to balance stem or precursor cell hierarchies. Silencing of genes such as p16, SFRPs, GATA4 and GATA5, and APC in the colon, for example, may lock these cells into a proliferative state, thus creating a population of cells at increased risk for additional epigenetic and genetic aberrations.
Recent epigenomic analyses have suggested that genes that control the growth and differentiation of stem cells are often suppressed by the polycomb repressive complex and that their activation results in the cessation of growth in stem cell populations. An early epigenetic alteration that could involve a switch from one type of silencing mechanism to another may be pivotal in moving the genes from a repressed but reversible configuration into a permanently locked state. Indeed, genes silenced by the polycomb repressive complex that involves H3K27me3 are at a much increased risk of switching to a DNA methylation state.16 Locking these genes may make it difficult for cells to express them at key developmental times in order to undergo differentiation. The change in silencing mechanism, however, makes genes sensitive to drugs that can unlock them and potentially restore normal cell regulation. If these epigenetic gatekeeper genes are at the root of the disruption of normal stem cell homeostasis in epithelia, epigenetic drugs may be able to reverse the early steps of carcinogenesis.
Despite the fact that we now know of many classes of genes that become inappropriately silenced during the formation of human cancer, we still do not fully understand the mechanisms. As mentioned above, genes subject to polycomb silencing in embryonic stem cells and other stem cell types seem to be particularly disposed to switch to DNA methylation silencing in cancer. Conversely, many of the genes that become silenced are not regulated by the polycomb system yet become methylated in cancer. Although some types of genes seem predisposed to silencing by DNA methylation, natural selection may play a role in the evolution of tumors in cells that have undergone epigenetic alterations. For example, epigenetic silencing of a gene (such as p16) that restricts cellular growth might allow for the progressive selection of cells that have gradually begun to silence the gene. Evidence for the role of selection comes from studies in which tumors harboring p16 gene mutations show expression of the mutant but not the wild-type allele.18 Since the promoters of both the wild-type and mutant allele are identical, yet only one undergoes DNA methylation silencing, epigenetic processes in cancer cells may be selected for in the host by giving a cell a growth advantage in an evolving process.19
The role of the polycomb repressive complex 2 (PRC2) in preventing cellular differentiation in embryonic stem cells and its subsequent dysregulation in cancer provides a basis for understanding these two processes. Levels of the histone methyltransferase (EZH2), which is part of the polycomb complex and another protein BMI1, are significantly up-regulated in cancer.20 The normal function of PRC2 is to control genes in embryonic stem cells and differentiated cells; up-regulation of these proteins may repress genes controlling cell division. The mechanisms responsible for the up-regulation of EZH2 have remained obscure; however, a particular miRNA (mir101) has recently been found to down-regulate the level of EZH2 in physiologic states.21 Since mir101 is commonly down-regulated in cancer, this may lead to an increase in the EZH2 protein followed by down-regulation of genes, decreasing differentiation and increasing cell growth. This type of dysregulation may be important in the regulation of stem cells and possibly cancer progenitor cells and thus represent an excellent drug target.
Inappropriate gene activation is also a hallmark of cancer, and this process can have an epigenetic basis as well. Hypomethylation of the genome is common in most human neoplasms and leads to demethylation of repetitive elements such as Alus and long interspersed nuclear elements (LINEs), resulting in genome instability. Alternatively it might lead to ectopic gene expression if it occurs in a potentially functional Alu or LINE promoter.22 Hypomethylation of CpG-poor promoters may also activate cancer-related genes such as oncogenes. Translocations may also reprogram epigenetic modifiers such as in the MLL fusion gene, which is a histone methyltransfease resulting in decreased levels of H3K4me3 (Table 3.1, fig. 3.1) and decreased gene expression.
EPIGENETIC BIOMARKERS FOR EARLY DETECTION OF CANCER
Since aberrant DNA hypermethylation is among the most common molecular alterations in human neoplasia and involved in the early stages of tumorigenesis, it lends itself as a biomarker for early detection with the ultimate goal of preventing advanced stages of cancer and death. Attributes of a good biomarker include high sensitivity and specificity. DNA hypermethylation of gatekeeper genes such as tumor-suppressor genes is highly specific to neoplastic cells, and methylation microarrays have revealed tissue- and tumor-type-specific patterns. A panel of genes with a high frequency of hypermethylation in cancer can probably be identified,23 and sensitive methods for the detection of DNA methylation are available. Easy accessibility of DNA from tumor cells is essential for the successful population-based application. As tumor tissue cannot always be easily obtained, sophisticated techniques are available to capture circulating tumor cells in blood, urine, and other bodily fluids24 may be coupled with highly sensitive DNA methylation methods. Alternatively, DNA from peripheral blood lymphocytes or whole blood cells has shown aberrant methylation correlated with patterns found in tumor tissues and is easily accessible.25
A number of studies support the potential of DNA methylation markers for the early detection of various cancers, including prostate,26 bladder,27 breast,28 lung,29 and others. Some of these studies have demonstrated that aberrant methylation changes can be observed several years prior to cancer diagnosis. Additional studies with improved design, including larger samples size, appropriate selection of the case and control population with prediagnostic samples, adjustment for confounding variables, and use of standardized DNA methylation techniques, will be necessary to establish and validate reference panels of characteristically methylated genes.30,31
EPIGENETIC THERAPIES
The occurrence of widespread epigenetic alterations, particularly aberrant DNA methylation in human cancers, has encouraged the development of drugs that can reverse these epimutations and restore normal gene expression patterns to cancer cells. The fact that epigenetic alterations can be observed early in the process of carcinogenesis makes this process an attractive target for chemoprevention. Currently there are four drugs that have been approved by the U.S. Food and Drug Administration (FDA) for the treatment of hematologic malignancies (Fig. 3.3). The nucleoside analogues 5-azacytidine (5-aza-CR) and 5-aza-2’-deoxycytidine (5-aza-CdR) were initially developed in Czechoslovakia in the 1960s as cancer chemotherapy agents.32 These drugs have unstable pyrimidine rings and it was thought that they might be effective cytotoxins following incorporation into DNA and the subsequent hydrolysis of the azanucleoside ring. The azanucleoside ring, however, is quite stable once incorporated into the DNA helix and acts as a powerful mechanism-based inhibitor of DNMTs.33 These enzymes, responsible for epigenetic maintenance of transcriptional memory, extract the cytosine ring from the DNA molecule, form a covalent bond with the six position of the pyrimidine, and reinsert the base into the helix following the transfer of a methyl group to the five position of the cytosine. DNMTs become attached to DNA containing 5-azacytosine, which leads to proteolytic destruction of the enzymes and a pharmacologic knockdown of DNMT activity.34 Thus, both drugs require incorporation into DNA, and the ribose analogue (5-aza-CR) needs to be reduced by ribonucleotide reductase in order to enter the DNA during DNA synthesis. Although both drugs are cytotoxic at high doses, their optimal biological effects in inducing gene expression are at lower concentrations. They are both extraordinarily effective at removing DNA methylation from newly synthesized DNA resulting in gene reactivation. Once a hypomethylation pattern has been induced, it can be carried over through subsequent cell divisions, resulting in prolonged alterations of gene expression. Unfortunately, cells show a tendency to remethylate DNA sequences that have been demethylated by drug treatment, resulting in the gradual blunting of the cellular response.35 This problem requires understanding of what triggers remethylation and the development of a new generation of therapeutics that can be repeatedly administered in order to prevent the remethylation process.
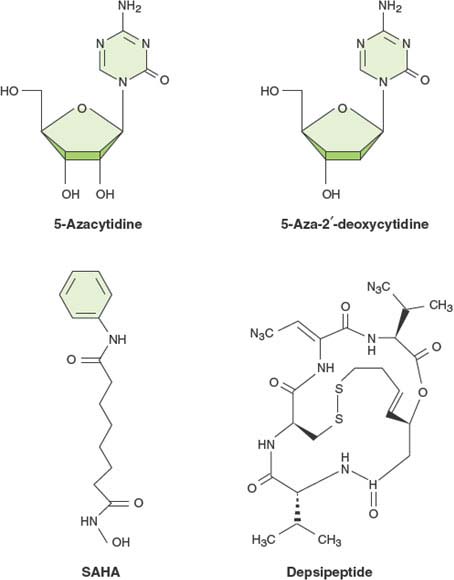
FIGURE 3.3 Structures of epigenetic drugs currently approved for use in humans by the U.S. Food and Drug Administration. The DNA methylation inhibitors (5-aza-CR and 5-aza-CdR) are both approved for use with myeloid dysplastic syndrome (MDS), and the two histone deacetylase inhibitors, suberoylanilide hydroxamic acid (SAHA) and depsipeptide, are approved for use in cutaneous T-cell lymphoma.
Although the causal relation between the chemically induced DNA hypomethylation and gene expression has been well established in cell culture models, it has been more difficult to demonstrate this causality in patients. The 5-Aza-CR prolonged life expectancy of myeloid dysplastic syndrome (MDS) patients in a phase 3 clinical trial36 and this hypomethylating agent has now become the standard of care for this type of premalignant condition. Global demethylation occurs in the cells of treated patients; however, the relation between the demethylation and a clinical response remains to be demonstrated.37 The deoxy analogue 5-aza-CdR (decitabine) has also shown good responses in a variety of small clinical trials; however, a large trial failed to show a survival benefit for patients.38 This result was surprising, since the deoxy analogue is more directly incorporated into DNA without concomitant incorporation into RNA and would therefore be expected to be more effective in the clinic. Possible explanations include a suboptimal study design regarding drug dosing and scheduling, and future trials will directly compare the efficacies of the two analogues.
The two histone deacetylase inhibitors suberoylanilide hydroxamic acid (SAHA) and depsipeptide have been found effective in the treatment of cutaneous T-cell lymphomas.39 These agents are relatively nonspecific inhibitors of histone deacetylase enzymes. As with the DNA demethylation agents, the clinical efficacy of the drugs have been demonstrated, yet the causal relation between histone acetylation and patient outcome remains to be shown.
Because epigenetic processes are highly interactive and reinforce one another, there is considerable interest in the development of combination therapies, in particular combinations between DNA demethylation and histone deacetylase inhibitors. Since gene expression requires both demethylation and histone acetylation, combinations of drugs that alter both processes may be more effective in treating cancer. Several clinical trials have been designed to test this hypothesis in an attempt to increase the reach of epigenetic therapies beyond the hematological arena into the treatment of solid tumors. Since many pathways relevant to cancer development are silenced simultaneously by epigenetic mechanisms, epigenetic therapy has the potential to reactivate them all at once (Fig. 3.4). The efficacy of epigenetic therapy may also be enhanced through combination with other treatment modalities (e.g., immunotherapy or chemotherapy).
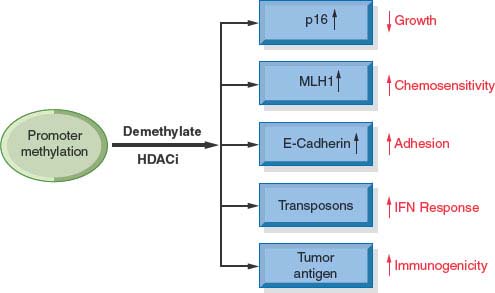
FIGURE 3.4 Combination therapies with DNA methylation inhibitors and histone deacetylase inhibitors can lead to the activation of genes inappropriately silenced in multiple pathways relevant for the generation of human cancer. Activation of these pathways by a drug regimen can result in many properties relevant to normal cell function such as decreased growth rate, increase in chemosensitivity, increased adhesion, and increased response to immunologic activities.
PROBLEMS WITH EPIGENETIC THERAPIES
Although epigenetic therapies have been clinically effective in randomized trials, problems remain with the widespread application to cancer care. One concern relates to the relative nonspecificity of the drugs in inhibiting epigenetic processes. For example, the DNMT inhibitors require incorporation into DNA where they are effective inhibitors of all three known DNMTs. Also, few specific histone deacetylase inhibitors are currently available. There is much interest in the development of more targeted drugs that may not require incorporation into DNA and be more specific in the enzymes they affect. Currently there are few inhibitors of other histone modifications such as H3K27me3, which is likely to be a critical target in cancer cells.
Another problem with current epigenetic therapies is their potential effects on normal cells and collateral damage to regular epigenetic processes within them, resulting in ectopic gene activation. However, since the DNA methylation inhibitors require incorporation into DNA to be effective, they have no measureable effects on noncycling cells, which constitute the bulk of the cell population within the patient. Conversely, cancer cells tend to scavenge nucleosides more effectively than normal cells, incorporating more drug into the DNA of cancer cells than normal cells, which mitigates some of the concern.40 There are also concerns related to the potential activation of oncogenic pathways in normal cells, although this does not appear to be as serious as initially imagined. However, patients currently receiving these drugs unfortunately do not have their lives extended to the point where secondary carcinogenic effects might become manifest. The remaining uncertainty makes it unlikely for these agents to find application in diseases that are not life-threatening. In the future, nonnucleoside inhibitors of DNA methylation that can be repeatedly administered may have the potential to reverse epimutations and significantly decrease the manifestation of cancer.
References
1. Jones PA, Liang G. Rethinking how DNA methylation patterns are maintained. Nat Rev Genet 2009;10:805.
2. Bernstein BE, Mikkelsen TS, Xie X, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006;125:315.
3. Campos EI, Reinberg D. Histones: annotating chromatin. Annu Rev Genet 2009;43:559.
4. Lin JC, Jeong S, Liang G, et al. Role of nucleosomal occupancy in the epigenetic silencing of the MLH1 CpG island. Cancer Cell 2007;12:432.
5. Riggs AD, Jones PA. 5-methylcytosine, gene regulation, and cancer. Adv Cancer Res 1983;40:1.
6. Jones PA, Laird PW. Cancer epigenetics comes of age. Nat Genet 1999;21:163.
7. Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A 1971;68:820.
8. Grady WM, Willis J, Guilford PJ, et al. Methylation of the CDH1 promoter as the second genetic hit in hereditary diffuse gastric cancer. Nat Genet 2000;26:16.
9. Toyota M, Ahuja N, Ohe-Toyota M, et al. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A 1999;96:8681.
10. Issa JP. Epigenetic variation and human disease. J Nutr 2002;132:2388S.
11. Christensen BC, Houseman EA, Marsit CJ, et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet 2009;5:e1000602.
12. Waterland RA, Michels KB. Epigenetic epidemiology of the developmental origins hypothesis. Annu Rev Nutr 2007; 27:363.
13. Marsit CJ, Houseman EA, Schned AR, et al. Promoter hypermethylation is associated with current smoking, age, gender and survival in bladder cancer. Carcinogenesis 2007;28:1745.
14. Michels KB. The promises and challenges of epigenetic epidemiology. Exp Gerontol 2010;45:297.
15. Niwa T, Tsukamoto T, Toyoda T, et al. Inflammatory processes triggered by Helicobacter pylori infection cause aberrant DNA methylation in gastric epithelial cells. Cancer Res 2010;70:1430.
16. Gal-Yam EN, Egger G, Iniguez L, et al. Frequent switching of polycomb repressive marks and DNA hypermethylation in the PC3 prostate cancer cell line. Proc Natl Acad Sci U S A 2008;105:12979.
17. Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet 2002;3:415.
18. Myohanen SK, Baylin SB, Herman JG. Hypermethylation can selectively silence individual p16ink4A alleles in neoplasia. Cancer Res 1998;58:591.
19. Varambally S, Cao Q, Mani RS, et al. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science 2008;322:1695.
20. Varambally S, Dhanasekaran SM, Zhou M, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002;419:624.
21. Friedman JM, Liang G, Liu CC, et al. The putative tumor suppressor microRNA-101 modulates the cancer epigenome by repressing the polycomb group protein EZH2. Cancer Res 2009;69:2623.
22. Wolff EM, Byun HM, Han HF, et al. Hypomethylation of a LINE-1 promoter activates an alternate transcript of the MET oncogene in bladders with cancer. PLoS Genet 2010;6 (4):e1000917.
23. Laird PW. The power and the promise of DNA methylation markers. Nat Rev Cancer 2003;3:253.
24. Nagrath S, Sequist LV, Maheswaran S, et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature 2007;450:1235.
25. Sharma G, Mirza S, Prasad CP, et al. Promoter hypermethylation of p16INK4A, p14ARF, CyclinD2 and Slit2 in serum and tumor DNA from breast cancer patients. Life Sci 2007; 80:1873.
26. Cairns P, Esteller M, Herman JG, et al. Molecular detection of prostate cancer in urine by GSTP1 hypermethylation. Clin Cancer Res 2001;7:2727.
27. Hoque MO, Begum S, Topaloglu O, et al. Quantitation of promoter methylation of multiple genes in urine DNA and bladder cancer detection. J Natl Cancer Inst 2006;98: 996.
28. Novak P, Jensen TJ, Garbe JC, et al. Stepwise DNA methylation changes are linked to escape from defined proliferation barriers and mammary epithelial cell immortalization. Cancer Res 2009;69:5251.
29. Palmisano WA, Divine KK, Saccomanno G, et al. Predicting lung cancer by detecting aberrant promoter methylation in sputum. Cancer Res 2000;60:5954.
30. Brooks J, Cairns P, Zeleniuch-Jacquotte A. Promoter methylation and the detection of breast cancer. Cancer Causes Control 2009;20:1539.
31. Cairns P. Gene methylation and early detection of genitourinary cancer: the road ahead. Nat Rev Cancer 2007;7:531.
32. Vesely J, Cihak A. 5-Azacytidine: mechanism of action and biological effects in mammalian cells. Pharmac Ther A 1978; 2:813.
33. Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell 1980;20:85.
34. Ghoshal K, Datta J, Majumder S, et al. 5-Aza-deoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology domain, and nuclear localization signal. Mol Cell Biol 2005;25:4727.
35. Bender CM, Gonzalgo ML, Gonzales FA, et al. Roles of cell division and gene transcription in the methylation of CpG islands. Mol Cell Biol 1999;19:6690.
36. Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol 2009;10:223.
37. Yang AS, Doshi KD, Choi SW, et al. DNA methylation changes after 5-aza-2’-deoxycytidine therapy in patients with leukemia. Cancer Res 2006;66:5495.
38. Lübbert M. Epigenetic therapy for myelodysplastic syndromes has entered center stage. Leuk Res 2009;33(Suppl 2):S27.
39. Marks PA, Xu WS. Histone deacetylase inhibitors: potential in cancer therapy. J Cell Biochem 2009;107:600.
40. Cheng JC, Yoo CB, Weisenberger DJ, et al. Preferential response of cancer cells to zebularine. Cancer Cell 2004;6: 151.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


