Cancer is a disease involving the failure of function of regulatory genes that control normal cellular homeostasis. The key roles of mutational processes in the generation of human cancer have been identified in the past decades. More recently the potential for epigenetic processes to complement genetic changes has been realized. In addition to multiple mutations, almost all human cancers contain substantial epigenetic abnormalities that cooperate with genetic lesions to generate the cancer phenotype. Epigenetic aberrations arise early in carcinogenesis preceding gene mutations and therefore provide targets for early detection. Epimutations may be reversed by drug treatments, providing the opportunity to design epigenetic therapies. This chapter will describe the role of epigenetic processes in cancer etiology and discuss their potential as biomarkers for early detection of cancer and precancerous lesions and their promise for drug development.
EPIGENETIC PROCESSES
Epigenetic processes are essential to ensure the appropriate packaging of the genome to fit within the confines of the mammalian nucleus, while maintaining its functionality. DNA is not found as a naked molecule in the nucleus but is wrapped up in nucleosomes composed of histone octamers and 146 base pairs (bp) of DNA, which are the fundamental building blocks of chromatin. Epigenetics is fundamental to organismal development: pluripotent cells arising at fertilization progressively lose their plasticity as they move through the consecutive differentiation steps necessary for embryogenesis. The recent development of whole epigenome approaches allows for the appreciation of the plethora of epigenomic processes that occur during development and the understanding of their role in activation and silencing of regulatory pathways.
The development of “next generation” sequencing approaches coupled with chromatin immunoprecipitation permits assessment of the distribution of the chemical “marks” imparted on the chromatin proteins and DNA. These epigenetic marks include DNA methylation and histone modifications (Table 3.1) and allow the orchestration of activation and silencing pathways. The marks or chemical modifications are placed on the chromatin components by enzymes such as methyltransferases and some of them can be removed by other enzymes (Table 3.1). While we are just beginning to understand the potential roles of specific chemical marks in ensuring the mitotically heritable variation in cell metabolism, which does not involve direct changes in the DNA sequence itself, the key role of a subset of these marks in controlling the potential for gene expression is becoming apparent (Table 3.1).
The fundamental process of DNA methylation applies methyl groups to cytosine residues in CpG dinucleotides to form 5-methylcytosine catalyzed by three DNA methyltransferase enzymes (DNMT1, DNMT3a, DNMT3b).1 Methylation patterns, once established, can be faithfully copied over a protracted period of time. The CpG dinucleotide is asymmetrically distributed in human DNA with about half of human genes containing CpG-rich regions termed “CpG islands” at their transcriptional start sites (TSS). Mostly, CpG islands are not methylated and genes are switched on or off without changing the methylation status of the CpG sites within islands. However, in certain physiologic situations such as X-chromosome inactivation or genomic imprinting, the CpG islands do become methylated in a manner that ensures permanent silencing due to the inherent mitotic heritability of the DNA methylation patterns. In contrast, embryonic stem cells keep genes quiet but poised for later expression during differentiation by using histone marks that are easier to reverse than DNA methylation to accomplish this purpose.2
The histone tails that protrude from the histone octamer, containing 146 bp of DNA in the nucleosome, are also modulated by enzymes and have functional significance for gene expression.3 Acetylation of the lysine residues (particularly lysines 9 and 14) is strongly associated with gene expression and is highly localized to the TSS of genes. The overall level of lysine modification in chromatin is dictated by opposing enzyme functions involving histone acetyltransferases (HATs) and histone deacetylases (HDACs), which apply or remove acetyl groups on lysine residues, respectively. The level of acetylation correlates with the level of expression, and HDACs have received considerable attention as potential drug targets. The TSS of human genes are also marked by the presence of three methyl groups on the lysine 4 residue of histone H3 (H3K4me3). Overexpression of enzymes that attach the methyl groups to this residue has profound implications for human cancer development. Trimethylation of histone H3 lysine 9 (H3K9me3) or lysine 27 (H3K27me3) is associated with gene repression (Table 3.1). The H3K9me3 is applied by several different methyltransferases, including G9a, and is associated with abnormally silenced methylated CpG islands. The H3K27me3 mark is applied by an enzyme of the polycomb repression complex 2, histone-lysine N-methyltransferase (EZH2), and aberrant activity of this enzyme is associated with human cancer development.
TABLE 3.1 SOME EPIGENETIC MODIFICATIONS AND THEIR ROLES IN GENE ACTIVITIES
None Histone deacetylase inhibitors (SAHA, depsipeptide) and many others
Histone H3
Lysine 4 methylation (H3K4me3)
MLL and several others
Gene activation
None
Histone H3
Lysine 9 methylation (H3K9me3)
G9a SUV39h
Gene repression
BIX-01294
Histone H3
Lysine 27 methylation (H3K27me3)
EZH2
Gene repression
3-deazaneplanocin A (DZNep)
Histone H2AZ
Replacement histone
Gene activation
None
The macromolecules that constitute chromatin undergo various covalent modifications, which result in cellular memory of transcriptional competency. The table lists only a subset of these marks and covalent modifications and focuses on those that are mainly localized to the start sites of human genes. The table also lists drugs that are currently in the clinic or are known to modify these processes, thus providing avenues for epigenetic therapies. DNMT, DNA methyltransferase; HAT, histone acetyltransferase; DHAC, histone deacetylases; SAHA, suberoylanilide hydroxamic acid; SUV39h, histone 3 lysine 9 trimethyltransferase; MLL, histone 3 lysine 4 trimethyltransferase; G9a, histone 3 lysine 9 trimethyltransferase, EZH2, histone 3 lysine 27 trimethyltransferase.
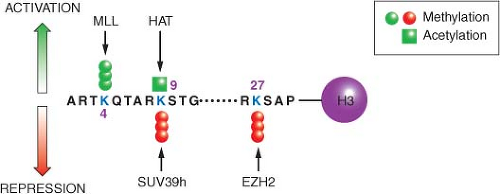
Figure 3.1 depicts the positions of a small subset of the possible modifications on the histone H3 protein in the context of nucleosomes. Although there are other modifications such as phosphorylation, ubiquitination, and sumolation of this and other histones, the discussion here is restricted to methylation and acetylation, since their function and potential for drug development is currently best understood. The various modifications can be interpreted by other proteins (not shown) sometimes called “readers,” which modify local chromatin structure to either stimulate or repress gene expression. Still other proteins (also not shown), such as histone deacetylases or histone demethylases, can remove the modifications in response to cellular and environmental signals, resulting in a dynamic state.
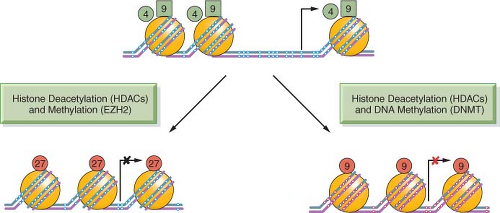
The positioning of the modifications relative to the TSS of genes is also critical for their function: Figure 3.2 shows the start site of a hypothetical gene with a CpG island in its promoter, which is normally free of DNA cytosine methylation. Active genes attach the activating H3K4me3 and H3K9ac modifications to the nucleosomes flanking the TSS. These may serve as “beacons,” allowing the transcriptional apparatus to “find” the start site and begin producing mRNA.
The silencing of the gene can be brought about in several ways, such as the removal of the activating H3K4me3 and H3K9ac modifications and application of the H3K27me3 mark (Fig. 3.2). Insertion of a nucleosome into the nucleosomefree region characteristic for CpG island-containing genes may also facilitate the silencing process. Genes silenced through DNA cytosine methylation also have nucleosomes at the start site of the gene but include the H3K9me3 modification rather than the H3K27me3 modification as a distinguishing feature.4 This more permanent mitotically heritable silencing process is used to ensure the long-term silencing of X-chromosome-linked genes and other important genes such as imprinted genes throughout the life of a human. These silencing mechanisms are all essential for mammalian development and maintenance of normal physiologic functions and can become pathologically altered in cancer and precancerous conditions, leading to widespread mitotically heritable aberrations in gene expression that characterize the cancer state.
FIGURE 3.1 Covalent modifications of histones can regulate gene activity. The location of activating and repressive marks on histone H3 are shown as an example. These covalent modifications, including trimethylation of lysine 4 (K4me3) and acetylation of lysine 9 (K9ac), are highly localized in start sites of genes and associated with active gene transcription. Conversely, methylation of lysine 9 (K9me3) or lysine 27 (K27me3) is associated with gene inactivity. It is the balance between these marks that define the transcriptional competence of a given gene. Unlike the two activating marks shown (green), the two repressive marks (red) tend to be more widely distributed on chromatin and potential drug targets. The figure is not intended to be comprehensive, and many additional modifications on histone H3 and other histones are known to participate in the structure of the epigenome. MLL, histone 3 lysine 4 trimethyltransferase; HAT, histone acetyltransferase; SUV39h, histone 3 lysine 9 trimethyltransferase; EZH2, histone 3 lysine 27 trimethyltransferase.
FIGURE 3.2 DNA methylation, histone modifications, and nucleosome occupancy define active and silenced states. The figure depicts a hypothetical CpG island that is active in gene expression and contains unmethylated CpG sites (white dots) within the CpG island and the presence of active histone H3 marks, including lysine 4 methylation (H3K4me3) (green circles) and lysine 9 acetylation (H3K9Ac) (green squares). Genes repressed by histone deacetylation and methylation by EZH2 show the insertion of a nucleosome (large orange circles) into the transcriptional start site and the application of lysine 27 trimethylation (H3K27me3) (red circles). This state is commonly observed in embryonic stem cells and holds genes in a poised silent state so that they can be called upon later for expression during embryogenesis and cell differentiation. The CpG island can also become silenced in a more permanent manner by histone deacetylation and DNA methylation, resulting in the presence of 5-methylcytosine (red dots) near the transcriptional start site. The histone mark associated with this state is often (but not exclusively) lysine 9 methylation (red circles). Genes silenced by this mechanism tend to be permanently silenced, and examples include genes on the inactive X-chromosome in mammalian cells.
Epigenetic processes such as those discussed above not only play important roles during development, but also are involved in maintaining tissue-specific patterns of gene expression in differentiated cells. In particular, DNA methylation patterns vary in different cell types, particularly at the transcription start sites of genes that do not contain CpG islands. Although the exact relations between these methylation patterns and control of gene expression have not been completely worked out, they probably assist in mitotic maintenance of differentiated states.
EPIGENOMIC CHANGES IN CANCER
DNA methylation patterns and histone modifications are essential for physiologic processes yet their dysregulation contributes to the cancer process. Epigenomic changes tend to be self-reinforcing and progressive and arise in normal cellular processes such as aging, which is one of the strongest risk factors for cancer. Several elements such as key genes that control the integrity of the genome (e.g., tumor suppressor genes, the adhesiveness of cells, the regulation of cell division, and the execution of apoptotic pathways) are all subject to inappropriate epigenetic silencing in cancer cells.
The field of cancer epigenetics started more than three decades ago with the observation that DNA methylation levels were profoundly altered in cancer cells relative to their normal counterparts.5 Subsequently, it became clear that the overall hypomethylation of the genome observed in cancer relative to normal cells was accompanied by focal hypermethylation near the TSS of key regulatory genes such as tumor suppressor genes.6 This focal hypermethylation of the CpG island regions at the TSS of the genes is associated with mitotically heritable silencing. Because of the inherent ability of DNA methylation patterns to be copied over a protracted time period, it was soon realized that these methylation changes could result in a molecular pathway that satisfies Knudson’s hypothesis for the inactivation of tumor suppressor genes. Knudson hypothesized that at least two hits were required for the inactivation of tumor suppressor genes in familial cancers such as retinoblastoma (RB).7 He proposed that mutations in the coding regions of genes such as the RB
Only gold members can continue reading. Log In or Register to continue