Summary of Key Points
- •
Distinct cellular phenotypes are based on differential gene expression, which is achieved through heritable epigenetic modifications that maintain active and inactive chromosomal regions.
- •
Epigenetic mechanisms include DNA methylation, histone modifications, regulatory DNA-binding proteins, regulatory RNAs, genome-organizing proteins, and chromatin remodeling complexes, all of which can be altered in lung cancer.
- •
Both genetic and epigenetic alterations can contribute to lung cancer and they can interact; genetic changes in epigenetic modifiers can affect the epigenome, and epigenetic silencing of genes involved in genome integrity can lead to genomic alterations.
- •
Numerous DNA methylation changes are seen in lung cancer, most commonly hypermethylation of promoter CpG-dense regions and loss of methylation in gene bodies, but only a fraction of DNA methylation alterations has functional consequences.
- •
DNA methylation alterations in a variety of bodily fluids can be used as biomarkers for the presence of lung cancer.
- •
Proteins involved in chromatin remodeling are commonly altered in lung cancer, with the ATPase BRG1 frequently mutated in non-small cell lung cancer tumors and cell lines.
- •
Epigenetic alterations are in principle reversible; epigenetic therapies thus offer opportunities for treatment by undoing cancer-driving epigenetic changes or by activating targets for therapy such as cancer/testis antigens and others.
Acknowledgments
DNA methylation research support for the Laird-Offringa lab comes from NIH/NCI grants R21 102247, R01 CA120689, R01 CA119029, the Canary Foundation, the Thomas G. Labrecque Foundation, the Whittier Foundation, and the Tobacco Disease-Related Research Program. Research support for chromatin remodeling in the Sanchez-Cespedes lab comes from the Spanish Ministry Economía y Competitividad (Spanish Grants SAF2011-22897 and RD12/0036/0045) and the European Community’s Seventh Framework Programme (FP7/2007-13), under grant agreement n°HEALTH-F2-2010-258677–CURELUNG. Neither the Laird-Offringa or the Sanchez-Cespedes laboratories accept any money from the tobacco industry. The content of this chapter is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
The cells that make up the human body exhibit an incredible variety of phenotypes, despite the fact that they all carry the same genome, inherited from a single fertilized egg. This phenotypic diversity arises from the different gene expression profiles in each distinct cell type and is achieved by creating and maintaining specific activated and inactivated genomic regions as cells differentiate into their destined types. These distinct genomic regions are established through the layering of information, or so-called biomarks, on top of the genome. The study of these regions and marks is called epigenetics.
Epigenetic information can come in many forms ( Fig. 12.1 ). One form is the direct chemical modification of DNA. The best studied chemical modification is DNA methylation, but more recently, hydroxymethylation, formylation, and carboxylation have also been noted. Chemical marks can also be deposited on the proteins that interact with DNA, the most prominent of which are the histones. Histones can be decorated with a wide variety of modifications, which can affect the accessibility of DNA to regulatory factors and thereby modulate the ability of genes to be expressed. In addition to covalent modifications of the histone tails, chromatin structure is also regulated by movement of nucleosomes in an adenosine triphosphate (ATP)–dependent manner through the activity of chromatin remodeling complexes. These complexes utilize ATP to disrupt nucleosome-DNA contacts and render DNA available to proteins requiring access to histones or DNA during distinct cellular processes. Besides histones and nucleosomes, other proteins can affect the epigenetic readout of the genome: numerous proteins or protein complexes bind directly or indirectly to the DNA and can either affect transcription through modulating the activity of enhancers or promoters, or can influence genomic organization and thereby gene activity. Lastly, regulatory RNAs, such as microRNAs (miRNAs) and long noncoding RNAs, exist that can epigenetically regulate gene expression. Collectively, these biomarks on the DNA are referred to as the epigenome; they are inherited following cell division, allowing cell phenotypes to be passed on to daughter cells.
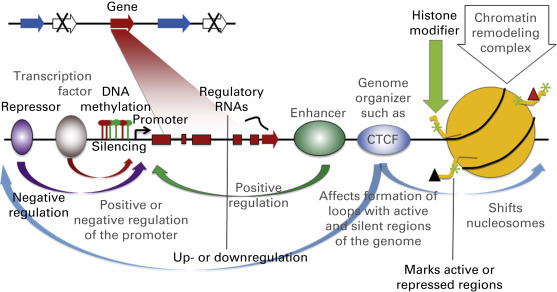
The importance of epigenetic marks in retaining proper cell phenotypes implies that their disruption would lead to disease. Indeed, it has become abundantly clear that epigenetic deregulation contributes very importantly to numerous diseases, including cancer. Epigenetic alterations have been widely implicated in the development and progression of lung cancer. Understanding the consequences of epigenetic changes can help dissect the molecular basis of lung cancer, providing insights into cancer development and progression, and thus new focal points for targeted therapies. In addition, epigenetic alterations in lung cancer show potential as molecular markers that could be applied to early detection, tumor classification, risk assessment, prognostication, and monitoring of cancer recurrence. Lastly, given that epigenetic information is layered on the genome without alteration of the DNA sequence, it is in principle reversible and is a prime target for the development and application of new therapies. Epigenetic drugs, such as histone deacetylase inhibitors and DNA methylation inhibitors, are in clinical trials for numerous cancers including those of the lung. With the advent of ever more powerful tools for genome-wide assessment of epigenetic marks, our understanding of the lung cancer epigenome and its application to diagnosis and treatment promises to increase dramatically in the years to come.
In this chapter, we review the basic concepts of epigenetics and discuss the current knowledge concerning epigenetic alterations in lung cancer, including the types of changes identified and their pathologic and clinical implications. Given the large number of epigenetic alterations analyzed to date and the dramatic acceleration in acquired data, it is impossible to be comprehensive in one chapter. Therefore, we discuss the basic principles and focus in more detail on two specific areas: chromatin remodeling and DNA methylation. These two examples beautifully illustrate the importance of considering the interplay between genetic and epigenetic alterations in cancer. Due to space limitations, reviews are cited throughout as a source of more detailed information.
Genetic and Epigenetic Interactions
Initial research into the molecular basis of lung cancer focused on genetic alterations, such as mutations, loss of heterozygosity, deletions, and gene amplification. Well-known examples of genetic alterations in lung cancer include mutations in V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS) , the epidermal growth factor receptor (EFGR) , and tumor protein 53 (TP53) . However, it has become abundantly clear that epigenetic alterations contribute equally importantly to the development and progression of lung cancer. Epigenetic alterations seen in lung cancer consist of changes in histone modifications, alterations in chromatin structure and chromatin-associated proteins, changes in regulatory RNAs such as microRNAs, and DNA methylation changes (both loss and gain of methylation).
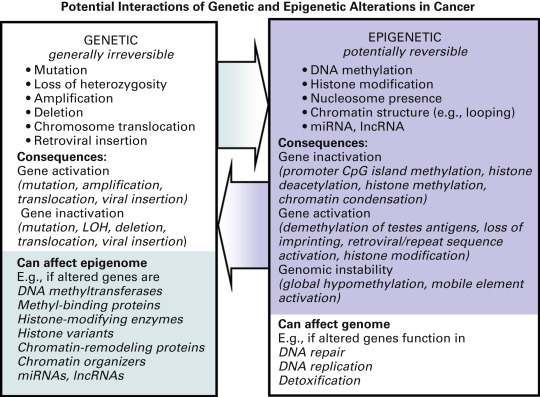
The interaction between genetic and epigenetic hits in cancer cells further amplifies the consequences of these molecular alterations ( Fig. 12.2 ). For example, as discussed later, genetic alterations in the genes encoding components of the epigenetic machinery (such as histone (de)acetylases, chromatin remodeling complexes, and DNA methyltransferases) can affect the activity of these enzymes and thereby the transcriptional activity of many additional genes. Somatic changes in parts of the epigenetic machinery are found in numerous cancers, including lung cancer. This potential for genetic alterations to affect epigenetics is further underscored by the reported link between genetic polymorphisms in several genes encoding epigenetic enzymes, and lung cancer risk. Conversely, epigenetic alterations can lead to further genetic damage. For example, hypermethylation of DNA repair genes or genes encoding detoxification enzymes can affect the cell’s susceptibility to mutagenesis and could result in the genetic (in)activation of additional genes. DNA methylation of 6- O -methylguanine DNA methyltransferase (MGMT) , an enzyme involved in the repair of alkylated guanine, is commonly seen in lung cancer. Inactivation of MGMT has been linked to an increase in the frequency of RAS gene mutation. In support of their potential to affect cancer development, polymorphisms in MGMT and other DNA repair genes have been linked to lung cancer risk in various populations. These examples illustrate that genetic and epigenetic changes should not be seen as independent, but rather as components of a complex interactive network that is responsible for the development and progression of numerous cancers, including lung cancer. Combined analysis of both types of molecular changes will accelerate the elucidation of the molecular pathways affected in lung cancer and may be especially helpful in characterizing particular types of lung cancer (e.g., histologic subtypes or lung cancer from smokers compared with nonsmokers). This holistic view of epigenetic alterations is also highly relevant to the clinic, as the use of certain cytotoxic drugs may potentiate or inhibit the efficacy of epigenetic drugs and vice versa.
Histone Modifications and their Role in Lung Cancer
The nucleosomal core around which DNA is coiled is composed of two molecules each of histones 2A, 2B, 3, and 4. The lysine and arginine-rich N -terminal regions extend from the core and can be heavily decorated with mono-, di-, and trimethylation, acetylation, ubiquitination, phosphorylation, and other modifications. These modifications do not exist in isolation; functional and physical crosstalk ensures a complex web of epigenetic signals, in which DNA methyltransferases, methyl-binding proteins, histone variants, histone modifying enzymes, and other chromatin and transcriptional components play a role ( Fig. 12.3 ). Many of the enzymes that modify histones recognize other modifications on the same or different histone tails or on DNA. For example, proteins that bind to methylated DNA frequently carry additional domains that interact directly or indirectly with histone-modifying proteins such as deacetylases. Acetylation of histones on lysine promote active transcription. On one hand, this modification reduces a positive charge and minimizes the electrostatic attraction of the histone tails for the DNA phosphate backbone, thereby relaxing chromatin structure. On the other hand, acetylated histone N -terminal tails are landing pads for bromodomain-containing proteins, such as transcriptional coactivator p300/CBP associated factor and TAF1, a component of the transcription initiation complex. Key acetylation marks are histone 3 lysine 27 acetylation (H3K27Ac), a mark found predominantly on active enhancers, and H3K9Ac, a mark found mainly on active promoters. Multiple enzymes that add or remove acetyl groups exist in the cell, and the deacetylases are particularly promising therapeutic targets in cancer. In addition to acetylation, common histone tail modifications are methylation, ubiquitination, and phosphorylation. Methylation does not affect histone tail charge, functioning instead by altering protein/protein interactions. One or two methyl groups can be added to arginine and up to three to lysine; the effects depend on the modified position and the number of added methyl groups. For example, histone 3 lysine 9 and lysine 27 trimethylation (H3K9me3, H3K27me3) are repressive marks, whereas H3K4me3 is found in transcribed regions.
As yet, relatively little is known about how histone modification is affected in lung cancer; molecular changes on the histone N -terminal regions are much more difficult to interrogate than DNA methylation changes. The most commonly used technique is chromatin immunoprecipitation (ChIP), in which formaldehyde crosslinking of cells is followed by specific immunoprecipitation of the proteins of interest (such as particular histone modifications) and local polymerase chain reaction (PCR)–based interrogation of specific regions. Due to advances in high-throughput sequencing technologies, ChIP can now be carried out genome-wide to gain global insights into the marks on or protein occupancy of the entire genome. In one study, researchers classified patients with non-small cell lung cancer (NSCLC) into seven distinct groups based on differential histone modifications and noted differences in survival depending on histology and histone 3 modifications. This early report hints at the potential use of this kind of epigenetic characterization to guide treatment. A key challenge in furthering these studies is the large amount of material needed for genome-wide interrogations, which limits current analyses largely to lung cancer cell lines. However, as the ability to extract epigenomic information from ever smaller quantities of material improves, the analysis of archival tumor specimens will become possible.
As alluded to previously, alterations in the enzymes that deposit or remove histone marks can lead to further epigenetic changes. A mutation of the histone acetyltransferase EP300 was found several years ago in a small cell lung cancer (SCLC) cell line. More recently, genome-wide sequencing approaches have provided further evidence for alterations in numerous genes encoding enzymes involved in histone modification ( Table 12.1 ). Mutations of the histone acetyltransferases CREBBP and EP300 and the histone methyltransferases MLL and MLL2 have now been detected in SCLC. In NSCLC, mutations in multiple histone methyltransferases including ASH1L, MLL3, MLL4, WHSC1L1, and SETD2 have been noted. Among these, one of the mutations in SETD2 was identified in a lung tumor from a never-smoker. More recently, amplification of the histone methyltransferase SETDB1 gene was found in a subset of NSCLC and SCLC cell lines and primary tumors. Depletion of SETDB1 expression in amplified cells was shown to reduce cancer growth in cell culture and in nude mice, whereas its overexpression increased tumor invasiveness. Mutations in histone demethylases and deacetylates have also been noted in NSCLC, further underlining the contributions of epigenetic deregulatory events in lung cancer.
| Enzyme | Lung Cancer Type | Reference(s) |
|---|---|---|
| Histone Demethylases | ||
| KDM6A | NSCLC | |
| Histone Methyltransferases | ||
| ASH1L | NSCLC | |
| MLL | NSCLC | |
| MLL2 | NSCLC | |
| MLL3 | NSCLC | |
| MLL4 | NSCLC | |
| SETD2 | NSCLC | |
| SETDB1 | NSCLC | |
| WHSC1L1 | NSCLC | |
| MLL | SCLC | |
| MLL2 | SCLC | |
| Histone Acetyltransferases | ||
| CREBBP | SCLC | |
| EP300 | SCLC | |
| Histone Deactylases | ||
| HDAC9 | NSCLC | |
| HDAC9 | NSCLC | |
a These data, obtained from high-throughput approaches, are preliminary, and validation in larger sets of well-characterized lung tumors is needed.
Abnormalities in genes encoding histone acetylases and deacetylases have also been found in noncancerous diseases of the lung, including chronic obstructive pulmonary disease (COPD), an irreversible and slowly progressive condition characterized by airflow limitation. Oxidative stress and inflammation are the major hallmarks of COPD, which, like lung cancer, has cigarette smoking as the major etiologic factor. In COPD, oxidative stress enhances inflammation by activating various kinase signaling pathways that lead to chromatin modifications (histone acetylation/deacetylation and histone methylation/demethylation). The activation of these pathways orchestrates several responses to stress, including proinflammatory and antioxidative responses. One of the main hurdles that precludes the clinical treatment of COPD is its resistance to antiinflammatory glucocorticoid (GC) treatment. GCs are not only involved in lung embryonic development and normal lung function but are also critical for lung cancer prevention. In this regard, a failure to respond to GCs constitutes a risk factor for developing lung cancer, especially in smokers. GC-mediated suppression of inflammation involves the recruitment by the GC receptor of histone deacetylase 2 (HDAC2) to the genes that mediate inflammation, resulting in histone deacetylation and reduced transcription. The GC-resistance in patients with COPD appears to occur as the result of a marked reduction in the levels and activity of HDAC2 in the lung parenchyma, provoked by the chemicals in cigarette smoke and by oxidative stress.
Taking into account that patients with bronchial obstructive changes, including COPD, are at increased risk for lung cancer, and that lung cancer cells are refractory to GC, it is interesting to speculate that the acquisition of resistance to GC may be among the factors that contribute to this increased lung cancer risk. However, there is as yet little evidence for decreased HDAC2 levels in lung cancer. One of the few reports examining the levels of HDACs in lung cancer specimens demonstrated a reduced expression of class II HDACs (HDACs 4–7, 9, and 10), especially HDAC10, and an association with poor prognosis. In contrast, a different study reported that increased expression of HDAC1 in tumor cells was an independent predictor of poor prognosis in patients with lung adenocarcinoma. Although the involvement of alterations in HDACs in lung cancer remains unclear, they should be considered as one of the possible mechanisms responsible for resistance to GC and similar compounds. Regardless of the possible involvement of alterations in HDACs, as discussed previously, loss of activity of chromatin remodeling complexes is among the most common causes of unresponsiveness to GC in lung cancer.
Chromatin Remodeling Complexes
Chromatin is controlled by multiprotein complexes that use the energy of ATP to disrupt histone–DNA contacts, thereby providing access to proteins that must contact DNA or histones during different cellular processes. These processes include the establishment of transcriptional control during embryonic development, cell differentiation, and the reprogramming of somatic cells, as well as the formation of heterochromatin, the execution of DNA repair, and DNA replication.
Four different families of chromatin remodeling complexes have been described: SWI/SNF, INO80, ISWI, and CHDs, distinguishable by the identity of their ATPase subunit. Several chromatin remodeling complexes have been implicated in cancer initiation and progression, most prominently the “switch/sucrose not fermenting” (SWI/SNF) complex. Therefore, we focus mainly on the SWI/SNF complex. However, it is anticipated that other chromatin remodeling complexes may also be ultimately found to be relevant in carcinogenesis. The name SWI/SNF describes genes first identified in yeast screens for mutants affecting mating type switching and sucrose fermenting. The SWI/SNF complex is powered by either of two ATPases that are highly homologous and have similar domains: ATP-dependent helicase SMARCA2 (BRM) or ATP-dependent helicase SMARCA4 (BRG1). In mammals, two different SWI/SNF multiprotein complexes have been described: BRG1 associated factors (BAF), also called SWI/SNF-α, and polybromo-BRG1 associated factors (PBAF), also known as SWI/SNF-β. The complexes contain a number of common proteins, such as BAF170, BAF155, BAF60a/b/c, BAF57, BAF53a/b, BAF47, BAF45a/b/c/d, and β-actin, but differ with respect to other components ( Fig. 12.4 ). The β complex contains up to three additional members: BAF180, BAF200, and BRD7. However, the main difference between the SWI/SNF complexes is the participating ATPase. In SWI/SNF-α the ATPase can be either BRG1 or BRM, but in SWI/SNF-β the ATPase is only BRG1. Numerous findings implicate subunits of these complexes in the development of lung cancer, as will be discussed. The nomenclature for involved factors is sometimes redundant ( Table 12.2 ).

| Name | |||||
|---|---|---|---|---|---|
| Official HNGC | Other Common | Other Less Common | Mutated in Cancer | Mutated in Lung Cancer | Characteristics of the Mutations in Lung Cancer |
| SMARCA4 | BRG1 | SNF2-like4, SNF2B, SNF2, SWI2, SNF2LB | Yes | Yes | More than 70% of truncating and nonsense mutations Mutations preferentially in NSCLC Mutations preferentially in smokers Mutations mutually exclusive with amplification of MYC and mutations of EGFR |
| ARID1A | SMARCF1, BAF250A | ELD, B120, OSA1, P270, hELD, BM029, MRD14 hOSA1, | Yes | Yes | About 50% of truncating and nonsense mutations Mutations preferentially in NSCLC |
| ARID2 | BAF200 | P200 | Yes | Yes | About 50% of truncating and nonsense mutations Mutations preferentially in NSCLC Mutations in smokers and nonsmokers |
| SMARCA2 | BRM | SNF2, SWI2, NCBRS, Sth1p, BAF190, SNF2L2 | Yes | NI | |
| SMARCB1 | INI1, SNF5, BAF47 | Snr1, MRD15, RTPS1, Sfh1p, hSNFS, SNF5L1 | Yes | No | |
| SMARCC1 | BAF155 | Rsc8, SRG3, SWI3, CRACC1 | Yes | NI | |
| ACTIN | ACTB | NI | NI | ||
| ACTL6A | BAF53A | ACTL6A | NI | NI | |
| ACTL6B | BAF53B | ACTL6B, | NI | NI | |
| ARID1B | BAF250B | OSA2, 6A3–5, DAN15, MRD12, P250R, BRIGHT | Yes | NI | |
| BRD7 | BP75; NAG4; CELTIX1 | Yes | NI | ||
| BRD9 | PRO9856; LAVS3040 | NI | NI | ||
| PBRM1 | BAF180 | PB1 | Yes | NI | |
| SMARCC2 | BAF170 | CRACC2, Rsc8 | NI | NI | |
| SMARCD1 | BAF60A | Rsc6p, CRACD1 | Yes | NI | |
| SMARCD2 | BAF60B | Rsc6p, CRACD2, PRO2451 | NI | NI | |
| SMARCD3 | BAF60C | Rsc6p, CRACD3 | NI | NI | |
| SMARCE1 | BAF57 | Yes | NI | ||
Genetic Alterations at SWI/SNF Chromatin Remodeling Factors in Lung Cancer
The first observation that linked chromatin remodeling and cancer development was the presence of inactivating mutations at SNF5 (also named SMARCB1 ) in rare cases of pediatric tumors, especially in malignant rhabdoid tumors. Mutations at SNF5 arise either somatically or in the germline, in the case of the germline conferring a cancer predisposition syndrome. Although SNF5 inactivation is infrequent in lung cancer, alterations at BRG1 (also named SMARCA4 ) are a more common event. Inactivation of BRG1 in lung cancer was first found by the detection of homozygous deletions of this gene in a small subset of cell lines of different tumor origins, including those from lung. The restoration of BRG1 in lung cancer cells induced growth arrest and a flattened phenotype. A few years later, it was reported that alterations at BRG1 occur in about one-third of NSCLC cell lines. A role of BRG1 loss in lung cancer has been verified in subsequent studies. Taking into account our current genetic knowledge of lung cancer, BRG1 can be considered among the five most commonly altered tumor suppressor genes in NSCLC, after TP53, CDKN2A, and LKB1 . In the vast majority of cases, the BRG1 mutations detected in lung cancer cell lines are biallelic and the mechanisms for inactivation include loss of one allele in combination with deletion of the other allele or frameshifts, indels, and nonsense/missense mutations, most of them yielding a truncated protein. The biallelic loss and inactivation mutations of BRG1 leave little doubt that BRG1 is a bona fide tumor suppressor gene, critical in the development of a large proportion of lung cancers. Mutations at BRG1 are more frequently detected in lung cancer cell lines as compared with lung primary tumors, similar to the findings for other tumor suppressor genes, such as TP53 , CDKN2A , and LKB1 . This finding could be attributed to technical difficulties related to the normal cell contamination often associated with the genetic analysis of primary tumors. In this regard, immunohistochemical evaluation of BRG1 levels in lung primary NSCLCs supports the absence of protein in 30% of the tumors. In contrast to BRG1 , genetic inactivation of BRM has not been reported in lung cancer. Some lung cancer cell lines have no detectable BRM protein by Western blot, but the mechanisms underlying the apparent loss of BRM protein are still elusive.
To date there is little information about the clinical, pathologic, and molecular correlations with the loss of BRG1 expression in lung cancer. Approximately one-third of NSCLC cell lines and 5% of SCLC cell lines have featured BRG1 inactivation. Moreover, these alterations seem to be associated with smoking and frequently coexist with mutations at other commonly altered genes in lung cancer, such as TP53 , KRAS , and LKB1 . However, it was noted that mutations in BRG1 were mutually exclusive with amplification of the V-Myc avian myelocytomatosis viral oncogene homolog (MYC) oncogenes. Although these findings are still preliminary, they indicate that MYC and BRG1 exert a similar biologic function during lung tumorigenesis, which is in agreement with functional observations demonstrating that the SWI/SNF complex is required for the CMYC -mediated gene transactivation and that recruitment of SWI/SNF to the promoters regulated by MYC depends on MYC-INI1 interaction. In further support of the MYC-BRG1 functional relationship in lung cancer cells, it was found that wild-type BRG1 is required to decrease the levels of the MYC protein in response to differentiation agents.
Immunohistochemical analysis of BRG1 in 41 primary lung adenocarcinomas and 19 primary lung squamous cell carcinomas showed that loss of nuclear expression of BRG1 and/or BRM was associated with worse survival in patients with NSCLC. In another study, the levels of several core proteins involved in the chromatin remodeling machinery were determined in 150 lung adenocarcinomas and 150 squamous cell carcinomas, using immunohistochemistry. Positive nuclear BRM staining correlated with a favorable prognosis in both patients with lung adenocarcinomas and primary lung squamous cell carcinomas with a 5-year survival rate of 53.5% compared with 32.3% for those whose tumors were negative for BRM ( p = 0.015). Furthermore, patients whose tumors stained positive for both BRM and BRG1 had a significantly better 5-year survival (72% compared with 33.6% for those whose tumors were positive for either or negative for both markers) ( p = 0.013). In another study, the authors found that loss of expression of BRG1 and BRM was found to be frequent in solid predominant lung adenocarcinomas and in tumors with low expression of lung transcription factor NKX2-1 (previously called thyroid transcription factor-1 or TTF-1) and low cytokeratins and E-cadherin. The same study demonstrated that the loss of BRG1 protein was mutually exclusive with EGFR mutations, which is in agreement with the higher frequency of BRG1 mutations in lung cancers in smokers ( Table 12.2 ).
In addition to mutations in BRG1 and BRM , novel deep sequencing technologies of lung cancer samples have allowed the identification of inactivating mutations in numerous genes encoding core members of the SWI/SNF complex, including ARID1A and ARID2 ( Table 12.2 ). Most of the data come from deep sequencing technologies applied to the whole genome or to exomes, a type of approach that leads to a certain number of false-negative results. It is anticipated that the rates of inactivation at each individual component of the SWI/SNF complex in the distinct types of tumors will become known in the coming years. Intriguingly, germline mutations of components of the SWI/SNF chromatin remodeling complex (i.e., SMARCB1, BRG1/SMARCA4, SMARCE1, ARID1A, ARID1B , and BRM/SMARCA2 ) have also been noted and they have been linked to human syndromes. In particular, the very rare autosomal dominant syndromes Nicolaides–Baraitser and Coffin–Siris show several common clinical characteristics, including multiple congenital malformations, microcephaly, and intellectual disability. To date, there is no information as to whether these syndromes confer a predisposition to lung cancer or any other type of cancer.
Although abnormal functioning of chromatin remodeling is now acknowledged to be involved in the development of lung cancer, our knowledge of the potential role of other epigenetic modifiers in lung cancer is still limited. As efforts to sequence the genomes of more tumors continue, more genes encoding chromatin modifiers are likely to emerge as mutated in lung cancer. However, functional analysis and validation in larger sets of well-characterized lung tumors will need to be undertaken to definitely determine the relevance of these different mutations in the development of lung cancer and to assess the precise frequency of alterations and the possible correlates with histopathology, etiology, and clinical parameters.
Functional Consequences of Abnormalities in Chromatin Remodelers and Other Epigenetic Modifiers in Lung Cancer Development
As previously mentioned, chromatin remodeling complexes function as transcriptional regulators of large sets of genes involved in multiple developmental pathways, including early embryonic development and tissue specification. The control exerted by the SWI/SNF complex on some of these processes is related to its involvement in regulating hormone-responsive promoters. Specific components of the SWI/SNF complex bind to various nuclear receptors for estrogens, progesterone, corticoids, retinoic acid, and vitamin D3, leading to their recruitment to gene-specific promoters. On the other hand, the ligands that bind specific nuclear receptors, such as retinoids and corticoids, are known to be critical for lung embryonic development as well as for normal lung differentiation and function. Given the involvement of the SWI/SNF complex in tissue specification and in developmental programs, one of the processes that would be expected to be disrupted in cancer cells carrying inactivated SWI/SNF is the capability to sustain gene expression programs during cell differentiation. Indeed, restoration of BRG1 activity in lung cancer cells induces gene expression changes, increasing resemblance to normal lung gene expression signatures. Furthermore, it was shown that lung cancer cells require wild-type BRG1 to respond to retinoic acid or GC treatment. Together, these findings support the notion that an inactive BRG1 confers resistance to retinoic acid and GCs, which prevent cancer cell differentiation. Given that BRG1 is part of the SWI/SNF complex, alterations in other members of the SWI/SNF complex may act through the same mechanism, although this hypothesis must be tested.
Several cancer-related proteins, such as P21, BRCA1, LKB1, SMADs, CFOS, CMYC, and FANCA, have been associated with BRG1 or other components of the SWI/SNF complex, supporting the participation of the SWI/SNF complex in other important cell processes, such as cell-cycle control, DNA repair, and the regulation of apoptosis in response to DNA damage. However, how the SWI/SNF complex affects these processes in the development of lung cancer is still poorly understood. A mouse model that permits conditional biallelic knockout of BRG1 in lung epithelial cells indicates that the loss of both BRG1 alleles induces apoptosis, as determined by significant increase in Apo-BrdUrd and cleaved caspase-3 in nontransformed lung epithelial cells. Furthermore, the homozygous loss of BRG1 in the lungs of the mice strongly potentiated the development of lung tumors after exposure to the carcinogen ethyl carbamate, which binds to DNA and generates DNA adducts. The observation of DNA adducts suggests that in the absence of a functional SWI/SNF complex, timely DNA repair is not guaranteed and, consequently, cells are inadequately protected from the deleterious consequences of DNA damage.
DNA Methylation
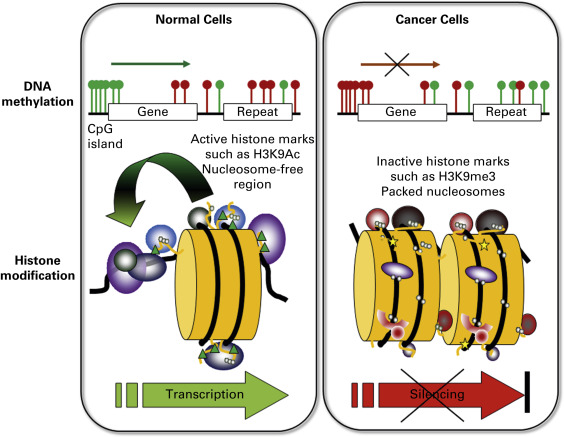
Of the covalent epigenetic marks, DNA methylation has been the most widely studied. The methyl group is deposited at the 5-position of cytosine, in the context of a small inverted repeat: a CpG dinucleotide. The palindromic nature of methylation allows the propagation of this modification following DNA replication. In the normal mammalian genome, some areas are heavily methylated, such as sections of the X chromosomes in females, pericentromeric regions, and parentally imprinted genes. Indeed, DNA methylation is essential for proper development and viability. Methylation in mammals is carried out by at least three enzymes: the maintenance DNA methyltransferase DNMT1 (which methylates daughter strands following DNA replication) and de novo DNA methyltransferases DNMT3A and 3B . All three genes are essential, as illustrated by mouse knock-out experiments. A large number of DNMT splice isoforms exist, a number of which appear to target particular genes or areas of the genome and some of which are implicated in cancer. Overexpression of DNMTs and the associated factor UHRF1 has been linked to lung cancer. CpG dinucleotides exist in two general environments in normal cells: sparsely distributed, and clustered ( Fig. 12.3 ). On the one hand, CpGs are sprinkled throughout the genome, and these CpGs are usually methylated. Spontaneous deamination of methyl-C results in thymine, which is less efficiently repaired than the uracil resulting from deamination of unmethylated cytosine. This has resulted in depletion over time of CpGs in areas that are usually methylated. Thus, the remaining dense clusters of CpGs, called CpG islands, are presumed to be normally unmethylated. It is estimated that about half of human genes contain such CpG islands in their promoter regions.
In cancer, a profound disruption of DNA methylation is seen ( Fig. 12.3 ). Broadly stated, global hypomethylation occurs, in combination with local hypermethylation. Loss of DNA methylation has been thought to contribute to carcinogenesis in two possible ways: the transcriptional activation of previously methylated sequences and the loss of chromosome stability. In contrast, the local hypermethylation at promoter CpG islands has been assumed to contribute to carcinogenesis through gene inactivation, silencing a wide variety of growth control and tumor suppressor genes, such as genes involved in proliferation, adhesion, apoptosis, the cell cycle, differentiation, signaling, and transcription. Although these findings remain generally true, genome-wide assessment of DNA methylation has painted a more complex picture, in which gain of DNA methylation can be paired with increased gene expression, and DNA methylation loss can be associated with repression. Genome-scale integration of DNA methylation and gene expression in lung cancer supports this observation. Of key relevance is the location of the alteration with respect to neighboring genes (promoter, intron, coding region) or gene-distant regulatory elements (enhancer, genome organizer), and whether DNA methylation changes affect the binding of regulatory factors.
One important reason for the extensive studies of DNA methylation to date is that relatively straightforward techniques exist to assess this modification. Our ability to analyze DNA methylation patterns has increased dramatically, from studies of single genes at a time in the 1990s to current genome-wide approaches. Given that methylation does not affect base pairing, and that DNA methylation information is lost following amplification by PCR, most DNA methylation assessing techniques employ a method to incorporate the DNA methylation information into the genomic sequence: bisulfite conversion. This process is a chemical treatment that converts unmethylated cytosines to uracil while methylated cytosines are protected; it allows methylation information to be incorporated into the DNA sequence. Bisulfite-converted DNA can be analyzed by many methods. Local bisulfite genomic sequencing, pyrosequencing, methylation-specific PCR, and its real-time version, MethyLight or its variation quantitative methyl-specific PCR, are still used to examine individual genes, but the development of increasingly powerful high-density bead arrays has provided a relatively low-cost way to interrogate hundreds of thousands of CpGs simultaneously. Bisulfite genomic sequencing originally consisted of amplification followed by cloning and sequencing, providing information on the methylation status of all the Cs on the same DNA strand in the amplified area. With the major advances in high-throughput sequencing, whole genome bisulfite sequencing has now become a reality, although it remains expensive and computationally challenging and, to our knowledge, has not yet been applied to entire lung cancer genomes. It is anticipated that the application of genome-wide methods will continue to provide ever more detailed insight into DNA methylation alterations in lung cancer. This knowledge promises to change the way in which lung cancer is detected and treated. The biggest challenge that lies in our future is the analysis and interpretation of the wealth of information obtained from genomic methods. Just as with mutations in cancer, some DNA methylation changes are drivers of the cancer process, while others are passengers, occurring coincidentally as molecular changes accumulate in cancer cells. Thus, identifying common DNA methylation changes in lung cancer is only the very beginning; determining which of these changes are cancer drivers is the much bigger task, one for which we have barely scratched the surface.
DNA Hypomethylation in Lung Cancer
Because of the overall hypomethylation found in cancer cells, researchers originally assumed that the cancer-causing effect of methylation changes was based on loss of promoter CpG island methylation resulting in proto-oncogene activation. Indeed, loss of methylation can promote the development of lung cancer in a murine model. Although genes can be activated by hypomethylation, they are not necessarily all considered canonical proto-oncogenes. One category of such genes is the parentally imprinted genes, genes for which either the maternally or paternally inherited allele is normally methylated. Hypomethylation can result in loss of imprinting, thereby contributing to cancer development; biallelic expression of the normally imprinted insulin-like growth factor 2, mesoderm-specific transcript, and H19 genes has been seen in lung cancer and is thought to contribute to the carcinogenic phenotype. Another type of gene that can be activated by hypomethylation is the family of testis-specific antigens; these genes are usually methylated and silent in all somatic tissues but the testes. Expression of testis-specific antigens has been noted in many tumor types including lung cancer, and these antigens are seen as potential immunotherapy targets. Loss of methylation of transposable elements and repeats is also found in lung cancer and can lead to mobility of such elements, causing further genetic damage. In addition, read-through from such demethylated elements may result in the aberrant activation of neighboring genes. Hypomethylation might also play a role in the activation of microRNAs, many of which are deregulated in cancer. For example, the normally methylated let-7a-3 microRNA was found to be hypomethylated in two of eight lung adenocarcinomas, and forced overexpression of this miRNA increased the oncogenic properties of lung cancer cell line A549.
In addition to contributing to carcinogenesis through gene activation, a second consequence of hypomethylation is thought to be genomic instability. Mice genetically engineered to underexpress DNA methyltransferases show an increased frequency of loss of heterozygosity and an elevated incidence of hematopoietic malignancies. Deletion of DNMT3A in a KRAS-dependent mouse of a lung cancer model was shown to promote tumor progression. However, analysis of methylation of five human squamous cell lung carcinomas and normal matched tissue showed prominent hypomethylation of repetitive elements but little methylation loss in single-copy sequences. This finding supports the notion that the effect of hypomethylation in lung cancer might be limited, which is important, because DNA methylation-blocking therapies are being widely explored, as will be discussed. These therapies appear to show promise in numerous cancers and are under investigation for lung cancer. Importantly, leukemia-prone DNMT underexpressing mice show a lower incidence of intestinal cancer, pointing to a protective effect of hypomethylation in certain tumor types. Indeed, treatment of a mouse xenograft model for human lung cancer with DNA methylation and histone deacetylation inhibitors suppressed tumor growth without apparent toxicity. A similar treatment of a murine lung cancer model cut lung tumor development in half, emphasizing the potential of epigenetic drugs for lung cancer treatment.
DNA Hypermethylation in Lung Cancer: Functional Implications
Although it would appear that the effects of hypomethylation in lung cancer are relatively modest, hypermethylation of promoter CpG islands is widely noted, with findings in lung cancer and DNA methylation described in more than 1500 reports in PubMed. Hypermethylation can be associated with transcriptional shutdown. This shutdown may happen directly, through steric interference of methylated cytosines with the binding sites for enhancer-binding proteins, transcription factors, cofactors, or chromatin organizers, or indirectly, through the attraction of methyl-binding proteins to the DNA, which in turn recruit histone deacetylase enzymes and other epigenetic modifiers ( Figs. 12.1 and 12.3 ).
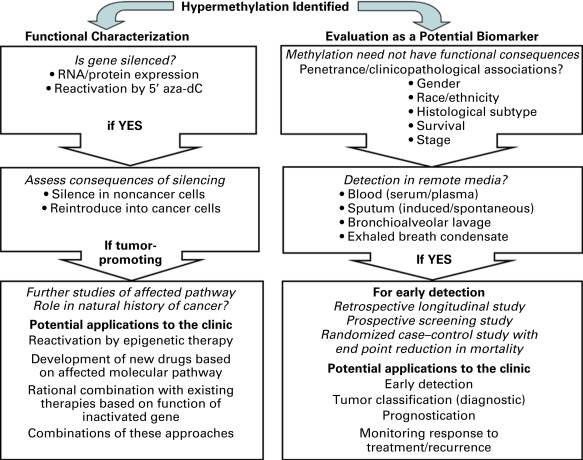
Intensive investigation will be required to determine whether DNA methylation changes are driver or passenger events in the development and progression of cancer. For the purposes of prognostication or providing tailored therapies, it could be of great importance to know whether DNA methylation events have functional consequences. This idea is supported by the prognostic utility of expression arrays. The first step in the approach to determine whether altered promoter CpG island methylation of a gene is functionally significant is the integration DNA methylation and gene expression information to identify genes showing concomitant changes in their DNA methylation and expression profiles ( Fig. 12.5 ). It has been frequently noted that only a minority of DNA methylation alterations show coincident changes in gene expression. This finding suggests that the rest are passenger events. However, there are other, as yet poorly explored, possibilities. For example, methylation changes may modulate binding of the chromatin organizer CTCF, thereby affecting alternative splicing events. Changes in splicing may not immediately be obvious from gene expression analyses, depending on the platform used to examine expression. Another option is that some of these DNA methylation alterations affect the expression of distant genes by modifying regulatory elements such as enhancers. Proper investigation of these possibilities will require more detailed genome-wide knowledge concerning alternative splicing patterns and investigation of epigenetic regulatory elements in the different types of lung epithelial cells that constitute lung cancer precursors. The analysis of trans-effects of DNA methylation on distant genes is computationally and biostatistically challenging and is only just beginning.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


