Epigenetic Abnormalities of Gastric Cancer
Annie On On Chan
Asif Rashid
INTRODUCTION
Epigenetics refers to changes in the phenotype (appearance) or expression of genes that are caused by mechanisms other than changes in the underlying DNA sequence. Neoplastic cells have three paradoxical changes in DNA methylation1: increased expression or activity of DNA methyltransferases,2 global hypomethylation,3 and de novo CpG-island methylation.1,2 Global reductions in the levels of 5-methylcytosine in a gene (hypomethylation) have been associated with tumorigenesis. Methylation of cytosine at CpG sequences is the only known DNA modification in human cells that can silence tumor suppressor genes in cancer. CpG methylation is an alternative to gene mutations and allelic losses as a mechanism for gene silencing, as demonstrated for a variety of genes, including the von Hippel-Lindau, human mut L homologue 1 (hMLH1) and p16 tumor suppressor genes.2 The primary evolutionary significance of DNA methylation has not been fully elucidated, but the leading hypothesis is that it evolved to silence and suppress the harmful effects of viruses and repeated DNA sequences.3 Methylation of cytosines within CpG islands is associated with loss of protein expression by repression of transcription and is observed in normal physiological conditions such as aging, X chromosome inactivation, and imprinting.1,2 CpG islands are 0.5- to 2-kilobase regions that are rich in the cytosine-guanine dinucleotides and present in the 5′ regions of about half of all human genes.4 Epigenetic silencing of tumor suppressor genes by CpG-island methylation is recognized as one of the most important mechanisms in carcinogenesis and is thought to initiate or act at an early stage of carcinogenesis. Gene silencing by CpG-island methylation is mediated in part by alterations in histone acetylation or methylation. The inactivation of chromatin by histone deacetylation suppresses transcription of tumor suppressor genes and likely also plays an early role in carcinogenesis.
Although decreasing in incidence, gastric cancer remains a major medical problem and is the second most common cause of cancer-related deaths worldwide. The incidence of gastric cancer is determined largely by environmental rather than genetic factors. The identification and subsequent classification of Helicobacter pylori infection as a type I carcinogen by the World Health Organization (WHO) were a major milestone in our understanding of the etiology of gastric cancer. Gastric cancer progresses through a series of morphological steps, from gastritis to early precancerous lesions to intestinal metaplasia and dysplasia to adenocarcinoma. The earliest alterations in the gastric mucosa involve epigenetic changes such as hypermethylation, which results in gene inactivation. This chapter focuses on the epigenetic changes along with environmental factors that contribute to gastric carcinogenesis.
CpG-ISLAND METHYLATION OF TUMOR SUPPRESSOR GENES IN GASTRIC CANCER AND NONNEOPLASTIC MUCOSAE
A large number of genes, including tumor suppressor genes, have been reported to be methylated in nonneoplastic mucosae, precursor lesions, and gastric cancers.5, 6, 7 and 8 Some of these genes include adenomatous polyposis coli, Cox-2, death-associated protein kinase, glutathione S-transferase P1, E-cadherin, hMLH1, O6-methylguanine methyltransferase, p14, p16, RAS association family 1A, thrombospondin 1, and tissue inhibitors of metalloproteinase 3. Methylation of these genes has been shown to accumulate in the multistep gastric carcinogenesis process5 and is associated with the increased expression of DNA methyltransferase 1 in gastric cancers.8
CpG-island methylation is dependent on the gene involved, the age of the patient, the underlying inflammatory conditions, and the site and histology of the nonneoplastic or neoplastic lesions. Similar patterns of age-related and cancer-specific methylation were observed in other epithelial malignancies besides gastric cancer.5 Some genes, such as death-associated protein kinase, are methylated with similar frequencies in nonneoplastic mucosae, precursor lesions, and invasive carcinomas.5 In contrast, other genes, such as the E-cadherin, p16, and hMLH1 tumor suppressor genes, show a stepwise increase in the frequency of methylation as the disease progresses from normal mucosa to invasive carcinoma.5,7 Methylation of some genes in nonneoplastic mucosae increases with the age of the patient and is more frequent in patients with gastric cancer than in those without gastric cancer.5 This age-associated increase in methylation in gastric nonneoplastic mucosae is observed for some genes, such as E-cadherin and p16, whereas others, such as hMLH1, show no increase in methylation.7
E-CADHERIN METHYLATION IN THE STOMACH
Of the genes that play a major role in gastric carcinogenesis, E-cadherin, a transmembrane glycoprotein that mediates cell adhesion, is one of the most well-studied.9 E-cadherin methylation has been reported in more than half of gastric cancers and is more common in diffuse-type carcinomas. Among patients with gastric cancers, those with intestinal metaplasia and invasive carcinomas have reduced expression of E-cadherin owing to E-cadherin methylation, suggesting that the loss of E-cadherin and its cell adhesion function plays a part in early gastric carcinogenesis.10 E-cadherin methylation has also been found in the nonneoplastic gastric mucosae of gastric cancer patients.7,10
Interestingly, E-cadherin methylation has also been found in nonneoplastic mucosae (without intestinal metaplasia) from one third of dyspepsia patients without gastric cancer and is associated with H. pylori infection.11 E-cadherin methylation has been found in the mucosae of approximately half of all patients with H. pylori infection (without intestinal metaplasia), and this methylation is reversible in at least two thirds of the patients when the infection is eradicated.11 In contrast, the methylation of other genes is not reversed by the eradication of H. pylori infection. Thus, it appears that H. pylori infection can induce reversible epigenetic alterations of the E-cadherin gene.
METHYLATION BY CHRONIC INFLAMMATORY CONDITIONS AND GASTRIC CARCINOGENESIS
CpG-island methylation is increased in patients with chronic inflammatory conditions and may contribute to the pathogenesis of tumors that develop in patients with these conditions. In the stomach, H. pylori and Epstein-Barr virus infections are associated with increased methylation in nonneoplastic mucosae, and studies have shown that increased CpG-island methylation in the
affected nonneoplastic mucosae is a precursor to gastric cancer.12,13 Thus, methylation may be one of the primary molecular mediators of gastric cancer and gastric cancer precursors arising in patients with inflammatory conditions, and this methylation is gene and precursor lesion dependent.
affected nonneoplastic mucosae is a precursor to gastric cancer.12,13 Thus, methylation may be one of the primary molecular mediators of gastric cancer and gastric cancer precursors arising in patients with inflammatory conditions, and this methylation is gene and precursor lesion dependent.
H. pylori infection also induces aberrant methylation in other genes and loci besides E-cadherin in nonneoplastic mucosae and precursor lesions.11,14 Methylation of eight CpG loci was higher in nonneoplastic mucosa of healthy volunteers with H. pylori infection or in nonneoplastic mucosa from gastric cancer patients without H. pylori
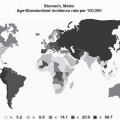 Epidemiology of Gastric Carcinoma
Epidemiology of Gastric Carcinoma
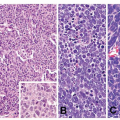 Neuroendocrine Tumors and Nonneoplastic Neuroendocrine Cell Changes
Neuroendocrine Tumors and Nonneoplastic Neuroendocrine Cell Changes
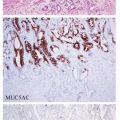 Gastric Carcinoma: Classifications and Morphologic Variants
Gastric Carcinoma: Classifications and Morphologic Variants
 New Imaging Techniques for the Diagnosis of Early Gastric Cancer and Premalignant Lesions
New Imaging Techniques for the Diagnosis of Early Gastric Cancer and Premalignant Lesions
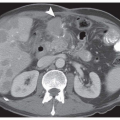 New Staging System for Gastric Cancer
New Staging System for Gastric Cancer
 Chronic Gastritis and Gastric Precancerous Conditions
Chronic Gastritis and Gastric Precancerous Conditions




Related posts:
Epidemiology of Gastric Carcinoma
Neuroendocrine Tumors and Nonneoplastic Neuroendocrine Cell Changes
Gastric Carcinoma: Classifications and Morphologic Variants
New Imaging Techniques for the Diagnosis of Early Gastric Cancer and Premalignant Lesions
New Staging System for Gastric Cancer
Chronic Gastritis and Gastric Precancerous Conditions
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
