Epidermal Growth Factor Receptor Inhibitors
Joel W. Neal
Lecia V. Sequist
The epidermal growth factor (EGF)-signaling pathway drives the growth and differentiation of epithelial cells. The epidermal growth factor receptor (EGFR) belongs to the ErbB family of transmembrane receptor tyrosine kinases and includes ErbB-1 (EGFR/Her1), ErbB-2 (HER2/neu), ErbB-3 (HER3), and ErbB-4 (HER4). EGFRstimulating ligands, including EGF, TGFα, amphiregulin, and epiregulin, bind to the extracellular domain of EGFR and induce receptor dimerization, leading to tyrosine phosphorylation of the intracellular domain by the tyrosine kinase domain of the receptors. Phosphorylated tyrosine residues attract signaling proteins and activate multiple downstream pathways, including the MAP kinase pathway, the PI3-K/Akt signaling pathway, and the JAK/STAT pathway.1 Many epithelial carcinomas have aberrant activation of EGFR signaling, which activates transcriptional programs leading to tumorigenesis, proliferation, survival, and angiogenesis.2
Two separate classes of drugs that target this pathway have demonstrated therapeutic effects in metastatic epithelial cancers, particularly non-small cell lung cancer (NSCLC), head and neck squamous cell cancer, colon cancer, and pancreatic cancer.3 The EGFR tyrosine kinase inhibitors erlotinib and gefitinib bind to the ATP-binding pocket of the intracellular kinase domain. This prevents the autophosphorylation of EGFR and subsequent downstream signaling effects. The monoclonal antibodies cetuximab and panitumumab bind specifically to the extracellular ligand-binding domain of EGFR. These appear to inhibit EGFR-dependent signaling through a variety of mechanisms, which include inhibition of ligand-dependent activation, inhibition of receptor dimerization and autophosphorylation, down-regulation of EGFR receptor expression, and induction of antibody-dependent cell-mediated cytotoxicity.3 Despite targeting the same pathway, these two classes of drugs appear to have dramatically different efficacies in different tumor types.
Gefitinib
The key features of gefitinib and erlotinib are shown in Table 27-1.
Chemistry
Gefitinib (ZD1839, IRESSA [4-(3-chloro-4-fluoroanilino)-7-methoxy-6-(3-morphiolenopropoxy)quinazoline]) is a low-molecular-weight quinazoline derivative. Gefitinib was identified through screens, which demonstrated inhibition of EGFR tyrosine kinase activity and the ability to kill EGFR-dependent cell lines. Structure-function relationships revealed that the quinazoline nucleus was required for activity, with electron-donating substituents at the 6- and 7-positions leading to increased potency.4 The chemical structure of gefitinib is displayed in Figure 27-1.
TABLE 27.1 Key features of gefitinib and erlotinib | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Mechanism of Action
Gefitinib inhibits the EGFR tyrosine kinase by virtue of competitive blockade of ATP binding. Gefitinib has an IC50 of 20 to 80 nM
for the wild type EGFR enzyme and is 100-fold less potent in vitro against related tyrosine kinases such as HER2 (ErbB2/neu). Gefitinib exhibits significant antitumor activity in a variety of human xenograft tumor models with high levels of EGFR expression.5
for the wild type EGFR enzyme and is 100-fold less potent in vitro against related tyrosine kinases such as HER2 (ErbB2/neu). Gefitinib exhibits significant antitumor activity in a variety of human xenograft tumor models with high levels of EGFR expression.5
 FIGURE 27-1 Chemical structure of gefitinib. |
Absorption, Distribution, and Elimination
Following oral administration of gefitinib, peak plasma concentrations are achieved within 3 to 7 hours, with a mean oral bioavailability of 60%. Administration of an oral dose of 225 mg daily results in steady-state levels of 200 ng/mL achieved after seven to ten doses. At steady state, circulating plasma concentrations typically are maintained within a twofold to threefold range over the 24-hour dosing interval. The volume of distribution is 1,400 L, and the mean terminal half-life is 41 hours. Elimination of gefitinib is primarily by hepatic CYP3A4 metabolism. The inactive metabolites are excreted in feces.6
Therapeutic Uses
Gefitinib was initially Federal Drug Administration (FDA) approved for the third-line treatment of patients with non-small cell lung cancer based on promising results in two phase II clinical trials.7,8 However, a phase III randomized, placebo-controlled trial of 1,692 patients compared gefitinib with best supportive care in the second-and third-line setting9 and failed to show a difference in overall survival, prompting the FDA to restrict gefitinib to patients who had previously received clinical benefit. Worldwide, gefitinib continues to be widely used in Asia, India and was recently approved for use in the European Union. Gefitinib has been tested in combination with chemotherapy in the first-line treatment of patients with NSCLC, but two large trials have failed to demonstrate a benefit with the addition of gefitinib to this regimen.10,11
While the effect of gefitinib was initially presumed to be due to the inhibition of wild-type EGFR signaling, around 10% of NSCLCs have mutations in the EGFR gene that confer exquisite sensitivity to this agent. Subgroup analysis of multiple clinical trials revealed that patients with adenocarcinoma with particular clinical features—never-smokers, Asians, and women—were significantly more likely to respond to gefitinib. Subsequently, it was revealed that tumors from patients with these clinical characteristics frequently have characteristic “sensitizing” mutations in EGFR.12 These mutations result in constitutive signaling through EGFR, leading to persistent activation of the MAP kinase and PI3K/Akt signaling pathway. These mutations can be classified into two groups: small in-frame deletions within exon 19 and a point mutation (L858R) (Fig. 27-2). Lung cancer cell lines containing these mutations have increased sensitivity to gefitinib, as compared with EGFR wild type cell lines (Fig. 27-2).
A number of prospective phase II clinical trials have reported a high response rate of 50% to 70% in patients with EGFR mutations following treatment with first-line gefitinib or erlotinib.13, 14, 15 However, it was unclear whether there was also a benefit in patients with the classical clinical characteristics without EGFR mutations. Therefore, a phase III trial conducted in Asia randomized 1,217 never-smoking and light-smoking patients with lung adenocarcinoma to first-line gefitinib versus chemotherapy. Patients with tumors containing an EGFR mutation demonstrated a 71% response rate to gefitinib, as compared with only 1% in patients without a mutation.16 Progression-free survival was also somewhat better for the gefitinib-treated group overall but starkly better in the gefitinib-treated subset of patients with EGFR mutations. Additionally, a preplanned subgroup analysis of the INTEREST trial, which randomized patients to third-line gefitinib versus docetaxel, showed significantly better response rates and progression-free survival in patients with EGFR mutations who received gefitinib.17 These results have prompted the European Union to recently approve gefitinib for use in patients with advanced NSCLC and EGFR mutations.
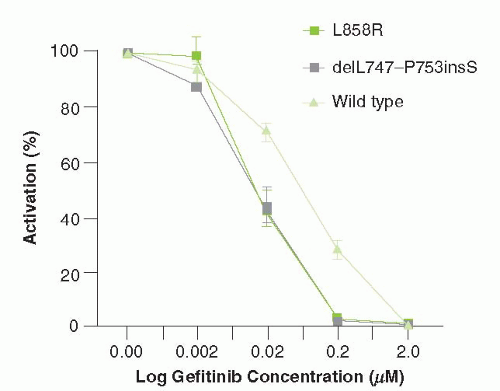 FIGURE 27-2 NSCLC cell lines containing mutations in EGFR are more sensitive to inhibition by gefitinib than wild type cells. Activation (Y-axis) is measured by degree of EGFR phosphotyrosine activation by immunoblotting. (Reproduced from Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004;350:2129-2139.) |
The standard dose of gefitinib is one 250-mg tablet daily, taken with or without food.
Adverse Effects; Drug Interactions
Diarrhea and pustular rash occur in approximately 50% of patients taking gefitinib. Other side effects include dry skin, nausea, vomiting, pruritus, anorexia, and fatigue. Most adverse effects occur within the 1st month of therapy and are tolerable when managed with supportive medications and dose reductions. Asymptomatic increases in liver transaminases have been observed, so periodic liver function testing should be performed. Interstitial lung disease, often associated with symptoms of cough and dyspnea, can occur acutely in 0.3% to 2% of patients receiving gefitinib, and up to one third of cases are fatal.6
Exposure to gefitinib is not significantly altered by food; however, drugs that cause sustained elevations in gastric pH to ≥5 reduce mean gefitinib area under the curve (AUC) by 47%. In vitro studies showed that the metabolism of gefitinib is predominantly via CYP3A4. Substances that induce CYP3A4 activity (e.g., phenytoin, carbamazepine, rifampin, barbiturates, or St. John’s wort) may increase metabolism and decrease gefitinib plasma concentrations and efficacy, and inhibitors of CYP3A4 (e.g., itraconazole, ketoconazole) may increase plasma concentrations. Patients using warfarin should be monitored closely for elevation of the international normalized ratio (INR) while taking gefitinib.
Erlotinib
Chemistry
Erlotinib (TARCEVA) is a quinazolinamine inhibitor of HER1/EGFR tyrosine kinase, with chemical formula N-(3-ethynylphenyl)-6, 7-bis(2-methoxyethoxy)-4-quinazolinamine. The chemical structure of erlotinib is displayed in Figure 27-3.
Mechanism of Action
Erlotinib is a potent inhibitor of the EGFR tyrosine kinase. Erlotinib competitively inhibits ATP binding in a manner similar to gefitinib. Erlotinib is more potent than gefitinib, with an IC50 of 2 nM.
Absorption, Distribution, and Excretion
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


