Fig. 25.1
Age-specific incidence rates of pancreatic cancer among men in the U.S., 2008–2012, by race/ethnicity. Data are taken from the U.S. SEER Registry
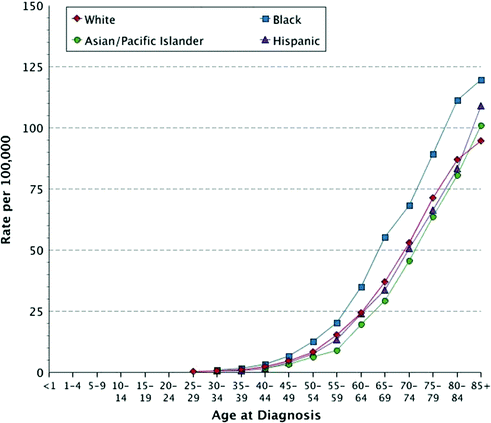
Fig. 25.2
Age-specific incidence rates of pancreatic cancer among women in the U.S., 2008–2012, by race/ethnicity. Data are taken from the U.S. SEER Registry
In the US, the 5-year survival rate is 7 % [2]. Mortality rates are very similar to incidence rates across all sexes and racial groups (age-adjusted rates 12.5 per 100,000 for men and 9.5 per 100,000 for women) [1].
In the US in 2015, an estimated 48,960 people will be diagnosed with pancreatic cancer and 40,560 will die of this disease [2]. Overall rates (incidence and mortality) for pancreatic cancer have been relatively constant over the past 3–4 decades, although a small increase in rates have been observed in the past decade [1]. Globally, pancreatic cancer incidence rates are higher in more developed countries particularly high rates are found in Eastern Europe, with the highest rates reported in Armenia, Hungary, Slovakia, and the Czech Republic (Globocan, 2015). An estimated 330,391 individuals die of pancreatic cancer annually in the world, accounting for 4 % of all cancer deaths. The majority of new cases are diagnosed in Asia (42 %), followed by Europe (30 %) and North America (14 %).
25.2 Risk Factors
25.2.1 Introduction
Our understanding of what causes pancreatic cancer improved dramatically over the past two decades as many prospective cohort studies achieved critical numbers of pancreatic cancer cases that allowed for more detailed analyses of suspected risk factors. In addition, a number of pooled and meta-analyses have been conducted to solidify strengths of associations. There now exists a large body of evidence for risk factors that are considered established risk factors of pancreatic cancer; these include family history, AB blood type, chronic pancreatitis, tobacco smoke, long-standing type 2 diabetes, high alcohol consumption, and obesity.
25.2.2 Tobacco Smoke
In a large pooled analysis from the International Pancreatic Cancer Case-Control Consortium (Panc4), 12 case-control studies were combined to include 6507 pancreatic cancer cases and 12,890 controls [3]. In this pooled analysis, the odds ratio (OR) for current smokers was 2.2, compared to never smokers, with a 95 % confidence interval (CI) of 1.7–2.8, which is consistent with results from prospective cohort studies where relative risks (RRs) have ranged between 1.7 and 2.5 [4–7]. Risk of pancreatic cancer increases with higher cigarette dose such that current smokers smoking 30 or more cigarettes per day, compared to never smokers, have a 2–3 fold higher risk [3, 4]. Former smokers have a much lower risk of pancreatic cancer; however, this estimate is largely dependent on time since smoking cessation among former smokers, as risk of pancreatic cancer nears that of never smokers 15–20 years after smoking cessation [3, 4]. Other cohort studies have reported shorter time periods after cessation (less than 10 years) for rates among former smokers to be similar to never smokers [6, 7].
Smoking cigars has also been associated with elevated pancreatic cancer risk for cigar smokers; the pooled OR from cigar-only smokers in a large case-control consortium (Panc4) was 1.62 (95 % CI 1.15–2.29), compared to never tobacco users [8]. In contrast, pipe smokers do not appear to have an increase risk in pancreatic cancer [8].
Measuring the association between smokeless tobacco (e.g., chewing tobacco/snuff) and pancreatic cancer poses a particular challenge, as confounding by smoking needs to be ruled out; consequently, the ideal populations to examine these associations are never smokers. Types of smokeless tobacco differ in the US and Europe and may have different impacts on risk. Three recent meta-analyses were performed on smokeless tobacco and cancer, but results for pancreatic cancer have been inconsistent [9–11]. A twofold increase in risk of pancreatic cancer among never smokers was reported in a retrospective cohort study of snuff users (RR 2.0, 95 % CI 1.2–3.3) [12], but another large cohort study did not report an increase in risk of pancreatic cancer among never smokers (only among smokers) [13]. At this time, the data on smokeless tobacco and pancreatic cancer are inconclusive.
For passive smoking, early exposure to cigarette smoke appears to increase risk of pancreatic cancer among never smokers, while environmental smoke from the workplace or home in adulthood do not appear to be associated with pancreatic cancer [14, 15]; other studies are needed to confirm these findings.
Examining mutation profiles in tumors may confirm associations and provide insight into mechanistic data to inform on causality. Mutations in the K-ras gene are common and arise early in the development of pancreatic cancer. To date, several studies have examined K-ras mutations in association with risk factors of pancreatic cancer, although they have often been limited by small number of cases with available tissue samples. In one study, smoking was associated with K-ras positive mutations in codon 12, but was not associated with K-ras negative tumors [16]. A positive association between smoking and K-ras mutations in pancreatic cancer was observed in two other studies [17, 18], but no associations were noted in three studies [19–21]. Recently, large mutational analysis (entailing sequencing of protein coding exons for >20,000 genes) compared pancreatic tumor tissue from smokers and nonsmokers; in this study, only one case (out of 114) did not have K-ras mutations [22]. Mutations in driver genes (KRAS, TP53, SMAD4, and CDKN2A/p16) were not more common in smokers, but smokers were observed to have more nonsynonymous mutations (p = 0.04) and more overall mutations (p = 0.06) than nonsmokers [22]. These findings suggest that smoking impacts pancreatic tissue in a way that is different from the lung tissue, as smoking has been strongly associated with K-ras mutations in lung tumor tissue [23, 24]. Lack of difference in mutations of driver genes suggest that smoking is not acting in the initiation events of pancreatic cancer. These findings are consistent with the observational data, given that smoking is not as strongly associated with pancreatic cancer as it is with lung cancer, and that a substantial risk reduction occurs after 10–15 years of smoking cessation. It is possible that smoking acts late in tumor development and through a number of nonspecific biological pathways.
25.2.3 Diabetes Type 2 and Hyperglycemia
It is well established that type 2 diabetes can develop as a consequence of pancreatic tumors and is often detected before the cancer diagnosis, but there has been substantial controversy over the role of diabetes as a cause of this cancer. There is considerable evidence supporting the role of glucose in the development of pancreatic cancer, both from epidemiologic and animal studies [25]. Prospective studies examining pre-diagnostic blood glucose levels have observed that individuals with elevated blood glucose a decade or more prior to diagnosis have a higher risk of pancreatic cancer [26]. The same observation has been made for long-standing type 2 diabetes; risk for pancreatic cancer remains elevated over 20 years post-diagnosis of diabetes type 2, and thus likely plays a causal role in carcinogenesis [27–29]. While a very high risk of pancreatic cancer is observed when diabetes type 2 is diagnosed within 1 year of the tumor diagnosis (RR 5.38, 95 % 3.49–8.30), a 50 % increase in risk is observed in individuals with a diabetes diagnosis 10 or more years before cancer diagnosis (RR 1.51, 95 % CI 1.16–1.96 [27]; RR 1.47, 95 % CI 0.94–2.31 [30]; RR 1.30, 95 % CI 1.03–1.63, compared to those without a diabetes diagnosis [28]) suggests that diabetes is both a consequence and cause of pancreatic cancer. Long-standing diabetes (>4 years) has also been associated with poorer survival after pancreatic cancer diagnosis [31].
In four prospective cohort studies, higher risk of pancreatic cancer was reported among participants with elevated blood glucose at baseline, compared to those with normal blood glucose [26, 32–34]. Two of these studies examined the temporal association between blood glucose and pancreatic cancer diagnosis [26, 34]. One study, including only nondiabetics, reported a statistically significant association for glucose levels above 198 mg/dL, compared to levels below 118.8, after excluding cases diagnosed in the first 5 years of follow-up (RR 1.97, 95 % CI 1.08–3.57) [34]. In the second study conducted among male smokers (ATBC study), risk remained elevated for those who had high blood glucose (>107 mg/dL), compared to <93 mg/dL, 10 or more years prior to diagnosis (RR 2.16, 95 % CI 1.05–4.42) [26].
In a large European cohort study, elevated glycated hemoglobin (HbA1c), a marker of prolonged elevated average glucose, was positively associated with pancreatic cancer risk (OR 2.42, 95 % CI 1.33–4.39, for highest [≥6.5 %, 48 mmol/mol] versus lowest [≤5.4 %, 36 mmol/mol] category) [35]. Risk with elevated HbA1C was slightly higher for those who were diagnosed 5 years or more after start of follow-up, suggesting that changes in glucose levels were not impacted by onset of disease. In another study pooling five prospective cohorts, HbA1c was also associated with risk of pancreatic cancer (OR = 1.79, 95 % CI = 1.17–2.72, comparing top to bottom quintile) and did not vary by time to diagnosis [36].
25.2.4 Obesity
Obesity has only recently been widely accepted as a risk factor for pancreatic cancer as many earlier case-control studies had not observed any associations for obesity. Over the past 15 years, prospective cohorts have published consistent positive associations for obesity and pancreatic cancer. To date, four meta-analyses and two pooled analyses of prospective cohort studies have been conducted examining body mass index (BMI) and pancreatic cancer risk. In the most recent and largest meta-analysis, which included 23 prospective studies and 9,504 pancreatic cancer cases, a 10 % increase in risk was reported for each 5-unit increase in BMI (RR 1.10, 95 % CI 1.07–1.14) [38]. A similar magnitude between obesity and pancreatic cancer was reported in other summary analyses (e.g., pooled study with 2170 cases of pancreatic cancer and 2209 matched controls: RR 1.13, 95 % CI 1.11–1.14 for a 5-unit increase in BMI [39]). The association between BMI and pancreatic cancer is not linear, however, and the increase in risk is more dramatic among very obese patients (RR 1.55, 95 % CI 1.16–2.07, comparing BMI >35 to BMI <25 kg/m2) [39]. Associations with obesity and pancreatic cancer are similar in men and women and by geographical region, but appear to be stronger among never smokers as the increase in risk is already apparent among those who were overweight [38].
Abdominal obesity (measured using waist circumference or waist-to-hip ratio) is also strongly associated with pancreatic cancer [38, 40]. The summary RR for a 10-cm increase in waist circumference was 1.11 (95 % CI 1.05–1.18) and for a 0.1-unit increment in waist-to-hip ratio was 1.19 (95 % CI 1.09–1.31) in the largest meta-analysis [38].
Obesity has also been associated with poorer survival from pancreatic cancer. The risk of death is roughly 50 % higher among patients with BMI >35 kg/m2 prior to diagnosis compared to those with normal BMI (<25 kg/m2) [41].
25.2.5 Chronic Pancreatitis
As with diabetes, pancreatitis is often diagnosed close to cancer diagnosis, making it difficult to disentangle cause and effect. The most convincing evidence for a causal role for chronic pancreatitis comes from studies on patients with hereditary pancreatitis. Symptoms of pancreatitis in patients with hereditary pancreatitis occur at a mean age of 10 years [42], and in these patients, rates of pancreatic cancer are not only much higher than those in the general population, but cancer develops at younger ages. In a national series study of 200 patients with hereditary pancreatitis (conducted in France), the average age at cancer onset was 55 years, and the standardized incidence ratio for this group, compared to the general population, was 87 (95 % CI, 42–113) [43]. Other hereditary pancreatitis case series, including two multi-site studies, reported slightly lower, but of similar magnitude, SIRs (53 [44] and 67 [45]). Smoking increases the risk of pancreatic cancer among those with hereditary pancreatitis and is associated with substantially earlier age at onset (50 years for ever smokers vs. 70 years for never smokers) [46]. As smoking prevalence was higher in the study conducted in France [43] (51 vs. 40 % in Lowenfels) [46], it may explain the higher relative risks noted in that study.
The incredibly high rates of pancreatic cancer observed among cases with hereditary pancreatitis, with known genetic mutations that lead to inflammation of the pancreas, provide support for a causal association between chronic pancreatitis (nonhereditary) and pancreatic cancer. The difficulty in studying chronic pancreatitis and pancreatic cancer risk includes small numbers, reverse causality, and confounding; studies that have examined the temporal relationship between chronic pancreatitis and diagnosis of pancreatic cancer find decreasing rates of cancer as the lag period increases [47]. As with type II diabetes, however, long-standing chronic pancreatitis is associated with a significant increase in risk; a 5.8-fold increase in risk of pancreatic cancer was estimated from six studies that excluded pancreatic cancer cases diagnosed within 2 years from chronic pancreatitis diagnosis (95 % CI 2.1–15.9) [47]. With a ten-year lag, the largest cohort study based on registry data reported a relative risk of 2.2, and although it was not statistically significant (95 % CI 0.9–4.4), only eight cases were available for this group [48].
Genetically modified pancreatic mouse models (developed to mimic human pancreatic adenocarcinoma) also support a role for chronic pancreatitis; induction of pancreatitis causes dramatic acceleration of pancreatic carcinoma in these mouse models [49]. These mouse models have provided many new insights into the mechanisms underlying pancreatic carcinogenesis and overwhelming evidence supports a role for inflammation [49].
25.2.6 Family History of Pancreatic Cancer and Genetic Susceptibility
Family history of pancreatic cancer explains a small fraction of pancreatic cases (<5 %); individuals with a parent, sibling, or child with pancreatic cancer have a moderately higher risk than those without family history (RR 1.76, 95 % CI 1.19–2.61) [50]. Family history of prostate cancer also appears to increase the risk of pancreatic cancer (RR 1.45, 95 % CI 1.12–1.89), but no associations were noted for those with family history of ovarian, breast, or colorectal cancers in one large study [50], despite the known elevated risk among those with BRCA2 mutations [51]. Germline mutations in BRCA2, CDKN2A, STK11, PRSS1, SPINK1, PRSS2, CTRC, and DNA mismatch repair have been associated with pancreatic cancer risk [52].
Genome-wide association studies (GWAS) have uncovered new areas of genetic susceptibility for pancreatic cancer. A region in the ABO gene (rs687289 at 9q34.2) was identified as being strongly associated with risk of pancreatic cancer in three GWAS analyses (PanScan III: ABO, OR 1.27, 95 % CI 1.20–1.35, P 1.6 × 10−16) [53–55]. These findings are consistent with studies that examined the role of blood groups directly, including suggestions for these associations as early as 1960 [56]. In a recent analysis, individuals with blood group A (HR 1.32, 95 % CI 1.02–1.72), AB (HR 1.51, 95 % CI 1.02–2.23), or B (HR 1.72, 95 % CI 1.25–2.38) had a higher risk of pancreatic cancer than those with blood group O [57]. Using these results, it was estimated that 17 % of the pancreatic cancer cases were attributable to inheriting a non-O blood group (blood group A, B, or AB) [57]. A number of other studies have confirmed the associations between blood type and pancreatic cancer risk [58–61].
In addition to ABO, three regions have been associated with pancreatic cancer risk using GWAS analyses (PanScan I and II) [53] and confirmed in PanScan III: rs9543325 at 13q22.1 (KLF5/KLF12, OR 1.23, 95 % CI 1.18–1.30), rs10919791 at 1q32.1 (NR5A2, OR 0.79, 95 % CI 0.75–0.85) and rs31490 at 5p15.33 (CLPTM1L, OR 1.20, 95 % CI 1.14–1.27) [55]. Recently, five new regions were identified in PanScan III: rs2736098 at 5p15.33 (a second signal in TERT), rs6971499 at 7q32.3 (LINC-PINT), rs7190458 at 16q23.1 (BCAR1/CTRB1/CTRB2), rs9581943 at 13q12.2 (PDX1), rs16986825 at 22q12.1 (ZNRF3) [55]. Several of the new loci identified in PanScan III are located in genes that have been implicated in pancreas development, pancreatic beta-cell function and predisposition to diabetes. While these findings provide insight into the pathways that are of importance for pancreatic carcinogenesis, the small effects observed for these susceptibility regions cannot (alone or combined) contribute predictive ability in determining who is at higher risk of pancreatic cancer [62].
25.2.7 Helicobacter pylori Infection
Positive associations between Helicobacter pylori and pancreatic cancer have been observed in some observational studies. The first report of an association between H. pylori and pancreatic cancer risk came from a case-control study with a small control group (n = 27); a twofold increase in risk was observed (OR 2.1, 95 % CI 1.09–4.05) [63]. In the Alpha-Tocopherol, Beta-Carotene Cancer Prevention study (ATBC), a prospective cohort study of male smokers, men with H. pylori antibodies or CagA-positive strains had about a twofold elevated risk of pancreatic cancer, compared to men who were seronegative for those antibodies (OR 1.87, 95 % CI 1.05–3.34; OR 2.01, 95 % CI 1.09–3.70, respectively) [64]. Findings from subsequent studies with large case numbers have been weaker and not statistically significant [60, 65–67]. Results from recent meta-analyses are inconsistent and largely influenced by authors’ decisions regarding which studies to include/exclude [68, 69]. Overall, the evidence for an association for H. pylori and pancreatic cancer, when examined critically, is weak.
25.2.8 Peptic Ulcers
A number of epidemiological studies have examined the relationship between peptic ulcers and risk of pancreatic cancer. Results from cohort studies with large number of pancreatic cancer cases and detailed information on type of peptic ulcers (i.e., gastric vs. duodenal) observed positive associations with gastric ulcers, but not duodenal ulcers [70, 71]. While H. pylori infections are associated with both types of peptic ulcers, gastric ulcers are associated with low acid production while duodenal ulcers are associated with hyperacidity; consequently, nitrosamine levels are higher in individuals with gastric ulcers and may explain the association with pancreatic cancer risk. Low acidity, however, also allows for the colonization of other bacteria, which may provide an opportunity for oral bacteria to move into the stomach and the gut.
25.2.9 Periodontal Disease
Periodontal disease is an inflammatory disease of the gums; with advanced disease, inflammation in the gums can lead to gum recession, soft tissue damage, bone loss and tooth loss (severe periodontitis) [72]. As with many chronic diseases, periodontal disease has multiple risk factors, including smoking and diabetes, and several bacteria have been linked to the severity and progression of periodontitis [72]. Porphyromonas gingivalis, a periodontal pathogen, has been extensively studied due to its unique ability to evade the immune response [73]. Positive associations between periodontitis and pancreatic cancer risk have been reported in three separate cohort studies [74–76]. In the National Health and Nutrition Examination Survey (NHANES) I Epidemiologic Follow-up Study, individuals with periodontitis at baseline had a higher risk of fatal pancreatic cancer compared to those with healthy periodontium (RR 1.77, 95 % CI 0.85–1.85), after controlling for age and sex [74]. A strong positive association between periodontal disease and pancreatic cancer was reported in a prospective cohort study of male health professionals [75]. Participants self-reported tooth loss and periodontal disease at baseline and were subsequently followed for 16 years. During that period, 216 cases of pancreatic cancer were newly diagnosed. After adjusting for age, smoking, diabetes, body mass index, and a number of other dietary factors, men with bone loss from periodontal disease had a 64 % higher risk of pancreatic cancer compared to those reporting no bone loss from periodontal disease. Among never smokers, a twofold increase in pancreatic cancer risk was observed (RR 2.09, 95 % CI 1.18–3.71), ruling out the possibility that the overall association was confounded by smoking. Furthermore, the association was stronger among dentists (RR 1.91, 95 % CI 1.31–2.78) who more accurately report history of periodontal disease [77]. In a recent analysis of the NHANES III data, a fourfold increase in risk of pancreatic cancer was observed among those with severe periodontitis, although the association was not statistically significant due to small numbers of cases (RR 4.56, 95 % CI 0.93–22.3) [76].
The association between antibodies to periodontal pathogens and risk of pancreatic cancer has been examined in a large European cohort (EPIC) [78] using blood samples stored on over 385,000 men and women at baseline (i.e., prior to disease). Using a nested case-control study design of 405 pancreatic cancer cases and 410 controls, a greater than twofold increase in risk of pancreatic cancer was observed among those with high levels of antibodies to a pathogenic strain of P. gingivalis (OR 2.38, 95 % CI 1.16–4.90, comparing >200 ng/ml vs. <200 ng/ml) after adjusting for known risk factors [78]. In the NHANES III cohort study, elevated antibodies to P. gingivalis (>69.1 EU, compared to less than 69.1) were associated with a threefold increase risk of orodigestive cancer mortality (RR 3.03, 95 % CI 0.99–9.31). Removing subjects with clinically apparent periodontal disease only decreased the association slightly (RR 2.25, 95 % CI 1.23–4.14). A separate examination of P. gingivalis with pancreatic cancer mortality could not be conducted in that study due to insufficient case numbers [76].
25.2.10 Allergies and Immune Response
A number of studies have examined the potential role of allergies in pancreatic cancer. A meta-analysis conducted in 2005 reported a 18 % reduction in risk of pancreatic cancer (OR 0.82, 95 % CI 0.68–0.99) [79]. In a multicenter case-control study, a 36 % decrease in risk was noted for individuals with a history of allergies (OR 0.64, 95 % CI 0.50–0.82) [80]. A more recent pooled analysis of 10 case-control studies, including 3567 cases and 9145 controls, harmonized the allergy variables to examine “any allergies” and reported a borderline statistically significant inverse association (OR 0.79, 95 % CI 0.62–1.00), controlling for known risk factors [81]. Statistically significant inverse associations were also found for hay fever and allergies to animals in the pooled analysis, while associations for asthma, eczema and allergies to food were weaker [81]. In a case-control study, 30–50 % reduced risk of pancreatic cancer were observed among subjects who had positive skin prick results for hay fever allergens, dust/mold, or animal allergens [82]; moreover, age of allergy onset did not impact associations, suggesting cancer was not likely causing allergy onset.
A 45 % lower risk of pancreatic cancer was reported in individuals with high levels of 22 oral antibodies, when compared with those individuals with overall lower levels of those antibodies, in a nested case-control study (OR 0.55, 95 % CI 0.36–0.83) [78]. Given that both allergies and high antibodies to bacteria are associated with a Th2 immune response, these findings support the hypothesis that the adaptive immune response may play an important role in pancreatic carcinogenesis.
25.2.11 Diet
A large number of studies have examined various aspects of diet in relation to pancreatic cancer risk, but overall, findings have been largely inconsistent or null. This section will focus on results from cohort studies, as case-control studies on diet are prone to bias, and have often reported findings that have not been reproduced in cohort studies.
25.2.11.1 Alcohol
Several large pooled analyses on alcohol intake and pancreatic cancer have been conducted which have provided strong data on dose-response relationships [83–85]. Results from a pooled analysis, two pooling data from prospective cohort studies [83, 84] and one from case-control studies [85], consistently indicate that alcohol is only a risk factor for pancreatic cancer at higher intake levels. In the largest pooling study, including over 5500 cases, a statistically significant elevated risk was observed for those consuming 6 or more drinks/day, compared to those consuming 0 to <1 drinks/day (OR 1.46, 95 % CI 1.16–1.83) [85]; no interaction was noted for the most common risk factors, including age, sex, race, and smoking status. In a pooling second study, a statistically significant 22 % increase in risk was observed among those consuming >30 g of alcohol/day (about three drinks/day), compared to nondrinkers [84]; however, numbers were too small to examine associations at higher intake levels. In a third pooling study, alcohol intake at >60 g/day (about 6 drinks/day) was associated with a nonsignificant increase in risk (OR 1.38, 95 % CI 0.86–2.23), compared to those who drank some alcohol (>0 to <5 g/day) [83]. The pooled OR for total alcohol for >30 g/day was similar to the other pooled analysis by Genkinger et al. (OR 1.23, 95 % CI 0.97–1.57) [84]. Finally, a large prospective cohort study (NIH-AARP), including 1,149 cases of pancreatic cancer, reported a 45 % increased risk among those consuming >3 drinks (95 % CI 1.17–1.80) and a 55 % increase in risk among those consuming 6 or more drinks of alcohol/day compared to >0 to <1 drinks/day (RR 1.55, 95 % CI 1.13–2.13, P-trend = 0.004) [86].
Based on these findings, no increase in risk is observed for moderate alcohol drinkers (i.e., 1–2 drinks/day), and an elevated risk is only observed among heavy drinkers, with increasing risk at higher levels. Two of the large studies also reported that liquor consumption was more strongly associated with risk than other alcoholic beverages [83, 86], while one study found wine to be more strongly associated with risk [85]. It is unclear if these are linked to behavioral patterns, or are markers for heavier alcoholic consumption.
25.2.11.2 Folate Intake
A pooled analysis of 14 prospective cohort studies with 2915 pancreatic cancer cases reported no association with folate intake (RR 1.06, 95 % CI 0.80–1.16, comparing highest vs. lowest quintile of total folate intake, i.e., dietary and supplement use) [87]. Similarly, no association was observed for dietary folate without supplements, or for supplemental use (RR 0.94, 95 % CI 0.73–1.22, for highest vs. lowest tertile of folic acid supplement use). Several meta-analyses have reported inverse associations between dietary folate intake and pancreatic cancer [88–90]; however, publication bias appears to be a major issue in these meta-analyses, as demonstrated in a forest plot of a recent meta-analysis [90] and suggested by the number of publications from cohort studies (6 published studies vs. 14 cohorts included in the pooling project). Furthermore, these meta-analyses included both case-control and cohort studies; summary estimates from cohort studies were weaker than summary estimates for case-control data [89, 90].
Studies using blood to measure folate levels have been inconsistent [91–93]. In the EPIC study, a U-shaped relationship was observed, with modest elevated risk at both ends of the spectrum of plasma folate levels [92]. An inverse association was observed in the ATBC study [93], while no statistically significant associations were noted in a nested case-control analysis of four prospective cohorts [91].
Taken together, these data do not support a dose-response between folate intake and pancreatic cancer and higher intake of folate may not result in lower risk of pancreatic cancer, as previously observed.
25.2.11.3 Fruits and Vegetables
Numerous case-control studies have reported inverse associations between fruit and vegetable intake and pancreatic cancer, but as with many other cancers, these findings have not been replicated in cohort studies. The largest pooled analysis of 14 cohort studies reported no association for fruit, vegetable, or total fruit and vegetable consumption and pancreatic cancer [94]. In this pooling project, no inverse associations were noted for individual fruit or vegetable consumption. Similarly, no associations were reported for fruit and vegetable consumption in a large European cohort study (EPIC) that was not included in the pooling study, with 555 pancreatic cancer cases [95]. Inverse associations for subgroups of vegetables have been observed in a couple of cohort studies, but the associations were observed for different subgroups and thus likely to be chance findings. It is important to note that meta-analyses on fruit and vegetable consumption including case-control studies are likely to provide misleading conclusions, given that case-control studies have consistently reported inverse findings, which are likely the result of selection and reporting biases. Moreover, a recent meta-analysis of cohort studies reporting statistically significant inverse associations for fruits and vegetables only included three cohort studies (out of >12 published studies) and is highly misrepresentative [96].
25.2.11.4 Meat Consumption
Meat consumption has been linked to pancreatic cancer in several cohort studies [97–102], although the type of meat associated with risk has not been entirely consistent across these studies, and three cohort studies reported no associations [103–105]. The inconsistency in findings for meat intake may be due to differences in cooking practices that can influence the level of carcinogens consumed. Cooking meats at high temperatures, or on an open fire, can result in production of compounds that are known carcinogens, such as heterocyclic amines (e.g., DiMelQx and MelQx) and polycyclic aromatic hydrocarbons (e.g., benzo(a)pyrene, BaP). Details on cooking practices for meats (e.g., doneness preferences and cooking method) have been incorporated in some cohort questionnaires to address this issue. Results from two large cohort studies with detailed cooking data reported significant positive associations with elevated levels of DiMelQx, MelQx and mutagenic activity, but not with BaP levels [97, 98]. In one study, consuming a high amount of meat cook at high temperatures was associated with a 52 % increase in risk of pancreatic cancer in men (top vs. bottom quintile of meat cooked at high temperature, RR 1.52, 95 % CI 1.12–2.06), although no association was observed in women [98]. In the second cohort, consuming very well-done red meat was associated with a 60 % increase in risk (RR 1.60, 95 % CI 1.01–2.54), compared to those consuming meat that is medium or rare [97]. Although only two cohort studies have examined cooking doneness, their findings are consistent with previous case-control studies [106, 107]. Moreover, the fact that most cohort studies did not report a positive association with processed meats (only one study observed a statistically significant association for processed meats [99]), but many reported positive associations with red meat, supports the possibility that cooking doneness plays a role in etiology.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


