Objectives
- 1.
Explain energy metabolism in general and how the challenges to provide adequate energy to all cells change during the digestive phase versus the fasting phase.
- 2.
List the primary hormones involved in the regulation of energy metabolic homeostasis, and describe their site of synthesis, regulation of production, and receptor signaling pathways.
- 3.
Diagram the hormonal regulation of specific enzymatic pathways in the hepatocyte, skeletal muscle fiber, and adipocyte during the digestive phase.
- 4.
Diagram the hormonal regulation of specific enzymatic pathways in the hepatocyte, skeletal muscle fiber, and adipocyte during the fasting phase.
- 5.
Explain the role of adipose tissue as an endocrine organ.
- 6.
Explain imbalances in energy metabolism and their consequences in type 1 and type 2 diabetes mellitus.
Overview of Energy Metabolism
Absolute, Continuous, and Universal Requirement for Adenosine Triphosphate
Cells continually perform work to grow, proliferate, and migrate; to maintain their structural integrity and internal environment; to respond to stimuli; and to perform their differentiated functions such as contraction, secretion, phagocytosis and propagation of an action potential. For example, while reading the previous sentence, your heart contracted and relaxed about 16 times, and about 3.5 million red blood cells entered your blood from bone marrow. Indeed, the resting metabolic rate (as when sitting and reading) constitutes about 60% to 70% of total potential energy expenditure.
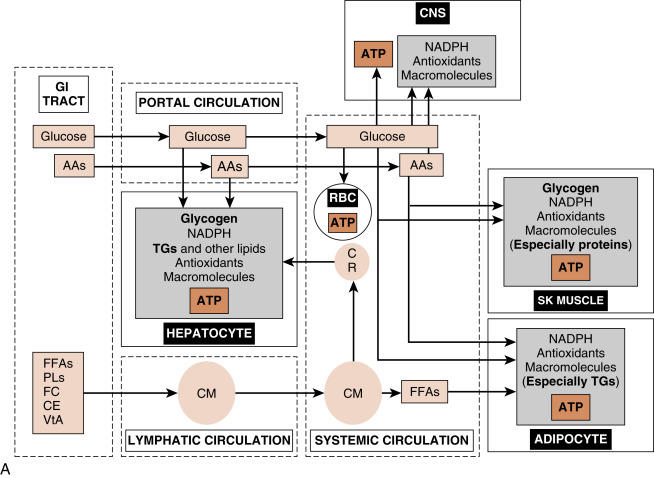
Cells derive their energy to perform this nonstop work primarily from the universal energy carrier, adenosine triphosphate (ATP) . The enzymatic hydrolysis of the terminal one or two phosphate groups releases a significant amount of energy that is coupled to and drives many energetically unfavorable reactions. However, ATP cannot be stored. Rather, cells need a continually renewable supply of ATP, which is achieved by continually oxidizing carbon-based fuels ( Figs. 3.1A and B ). The primary, monomeric fuels are monosaccharides (primarily glucose) , free fatty acids (FFAs ; also called nonesterified fatty acids), amino acids (AAs), and ketone bodies . Certain cells can store some of these fuels as polymers and access them as needed (e.g., skeletal muscle stores glucose as glycogen to be used during exercise). However, most cell types continually import monomeric fuels that are delivered to the extracellular microenvironment from the circulation and then enter cells via specific transporters.
Because cells depend on the continuous delivery of monomeric fuels from the circulation, the body as a whole must (1) maintain adequate circulation to all cells and (2) maintain adequate levels of primary fuels and O 2 within the circulation at all times. Inability to do so results in reversible cell injury and, ultimately, irreversible cell injury and cell death. Conversely, while inadequate levels of fuels can lead to cell dysfunction and death, excessive levels of fuels, both extracellularly and intracellularly, can also lead to serious injury (see later). Thus the regulation of whole body energy metabolism serves to maintain adequate levels of intracellular ATP in all cell types at all times, while keeping the concentrations of intracellular and circulating fuels below critical thresholds. The amount and type of circulating fuels available to cells change during the day, as well as during exercise, fasting, or starvation. Consequently, whole body metabolism needs to be dynamic and flexible. This is achieved through regulation of metabolism by endocrine, paracrine , and neuronal signaling , and by intracellular components that act as sensors of specific nutrients, metabolites, ions, or the adenosine monophosphate (AMP)/ATP ratio.
Cell-Specific Metabolism of Primary Fuels
For a specific fuel to be utilized by a specific cell type, that cell must express (1 ) cell membrane–associated transporters and, in some cases, intracellular organelle–associated transporters , that can import and export a specific fuel; and (2) the enzymes and cofactors that comprise specific metabolic pathways for a given fuel. Key components of these are regulated by hormones, metabolites, and intracellular energy sensors during the digestive versus fasting phases.
The cell types that play major roles in the coordination of fuel utilization and are major targets of hormonal and neuronal regulation are (1) the hepatocyte , (2) skeletal muscle fiber , and (3) the adipocyte (see Fig. 3.1A and B ). After the introduction of insulin and glucagon synthesis and regulation, this chapter will discuss the metabolic function and its regulation for each of these cell types in the context of the digestive phase and the fasting phase .
Nutrient Partitioning: The Digestive Phase
The digestive phase (or fed state) refers to the period of about 2 to 3 hours after the ingestion of a meal. Circulating fuels typically rise to levels that exceed the metabolic needs of the body. Because human metabolism has evolved to be very thrifty, excessive fuels are not wasted, but rather partitioned into various storage depots. The storage form of glucose is glycogen , the storage form of FFAs is triglyceride (TG) , and the storage form of AAs is protein , especially skeletal muscle proteins (see Fig. 3.1A ).
A major challenge associated with the digestive phase is to prevent prolonged excessive circulating and intracellular levels of nutrients . Insulin is the primary digestive phase hormone that promotes the movement of fuels out of the circulation and into cells, and their subsequent oxidation for energy or polymerization for storage or, in the case of AAs, for the synthesis of functional proteins. Insulin also promotes anabolic pathways that consume carbons from glucose, FFAs, and AAs, as well as consuming ATP, to build macromolecules to maintain cellular integrity and viability, or to promote cell growth and proliferation.
Nutrient Partitioning: The Fasting Phase
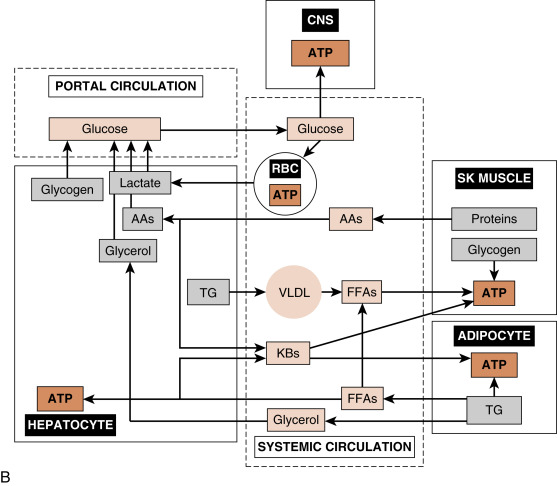
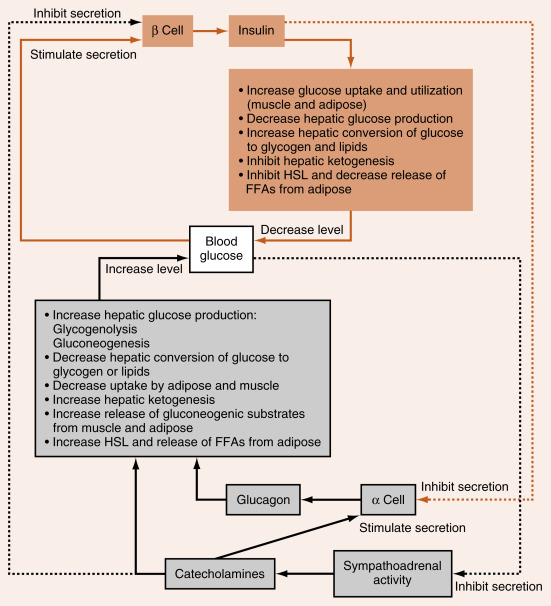
During the interdigestive phase between meals, the fasting phase during sleep and daytime fasting, and during prolonged physical work or exercise, the levels of circulating fuels begin to decline. In a normal, nonprotracted fasting phase (i.e., 8 to 12 hours), the brain is absolutely dependent on blood glucose for normal function. Thus a major challenge associated with the fasting phase is the maintenance of blood glucose levels above a critical threshold (60 to 70 mg/dL). This is required to avoid autonomic responses (e.g., nausea, heart racing), neuroglycopenic symptoms (e.g., confusion, lethargy), and even coma and death, due to increasingly severe degrees of hypoglycemia. Blood glucose levels are maintained by two processes (see Fig. 3.1B ):
- 1.
Hepatic glucose production. During the initial period of a fast, the liver breaks down stored glycogen via glycogenolysis to glucose-6-phosphate (G6P), converts G6P to glucose by the liver-specific enzyme, glucose-6-phosphatase, and exports glucose through the bidirectional GLUT2 transporter. As a fasting phase progresses, hepatic glucose production switches from primarily glycogenolysis to primarily gluconeogenesis, or the production of glucose from 3-carbon metabolites, including lactate, pyruvate, glycerol, and several “glucogenic” AAs that are released from proteolysis.
- 2.
Glucose sparing. Most cell types can utilize fuels other than glucose, thereby sparing glucose for use by the central nervous system (CNS). First, low levels of insulin during the fasting phase minimize the flow of glucose into skeletal muscle and adipocytes. TGs within adipocytes are mobilized so that FFAs are released into the circulation as FFAs, and preferentially used by skeletal and cardiac muscle, adipocytes, and other cell types for ATP production. Also, during the fasting phase, the liver converts FFAs and some “ketogenic” AAs into ketone bodies, and exports these fuels into the circulation for use by extrahepatic tissues. During a prolonged fast (i.e., 1 to 2 weeks), the CNS itself switches to the utilization of ketone bodies. Finally, skeletal muscle stores glycogen, which is mobilized especially during exercise/work for use within the muscle fiber.
The hormones glucagon and epinephrine , along with the sympathetic neurotransmitter, norepinephrine , represent the primary signals that induce the mobilization of energy stores and new synthesis of glucose and ketone bodies during the fasting phase, thereby preventing blood glucose from declining to dangerously low levels (see later). Several other hormones, including cortisol and growth hormone , also play important roles in the mobilization of energy stores, the control of circulating glucose and lipids, and the balance between protein synthesis and degradation; these are discussed in Chapters 5 and 7 .
Key Hormones Involved in Metabolic Homeostasis
Endocrine Pancreas
The islets of Langerhans constitute the endocrine portion of the pancreas (also called the endocrine pancreas ; Fig. 3.2 ). About 1 million islets, making up about 1% to 2% of total pancreatic mass, are spread throughout the pancreas. The islets are composed of several cell types, each producing a different hormone. In islets situated in the body, tail, and anterior portion of the head of the pancreas (all of which have a common embryologic origin), the most abundant cell type is the β cell . The β cells make up about three-fourths of islet cells and produce the hormone insulin . The α cells comprise about 10% of these islets and secrete the hormone glucagon . The third major cell type of the islets within these regions is the Δ cells , which make up about 5% of the cells and produce the peptide somatostatin (gastric somatostatin was discussed in Chapter 2 , as an inhibitor of gastrin secretion). A fourth cell type, the F cell, represents about 80% of the cells in the islets situated within the posterior portion of the head of the pancreas (including the uncinate process) and secretes the peptide, pancreatic polypeptide .
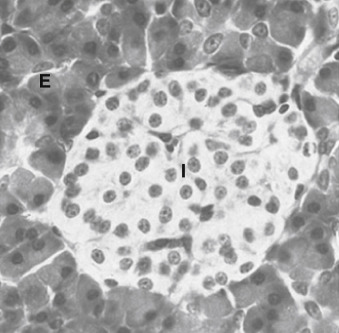
Because the physiologic function of pancreatic polypeptide in humans remains obscure, it is not further discussed here. Blood flow through the islets is somewhat autonomous from the blood flow to the surrounding exocrine tissue. Insulin secreted from the inner β cells reaches the outer α cells. Consequently, the first cells affected by circulating insulin are the α cells, in which insulin inhibits glucagon secretion.
Insulin
Insulin is the primary anabolic hormone that is responsible for maintaining the upper limit of blood glucose and FFA levels. Insulin achieves this by the following mechanisms:
- 1.
Increasing glucose uptake by skeletal muscle and adipocytes.
- 2.
Increasing metabolism of glucose in liver, skeletal muscle, and adipocytes.
- 3.
Increasing glycogen storage in liver and skeletal muscle.
- 4.
Increasing the clearance of chylomicrons from the blood.
- 5.
Increasing TG synthesis and storage in the liver and adipose tissue.
- 6.
Suppressing glucose output by the liver.
- 7.
Suppressing lipolysis of adipose TG stores.
- 8.
Suppressing very-low-density lipoprotein (VLDL) production by the liver.
Insulin also promotes protein synthesis from AAs and inhibits protein degradation in peripheral tissues. Finally, insulin regulates metabolic homeostasis through effects on satiety.
Insulin Structure, Synthesis, and Secretion
Insulin is a protein hormone that belongs to a gene family that also includes insulin-like growth factors I and II (IGF-I, IGF-II), relaxin , and several insulin-like peptides. Organized, functional islets appear in the human pancreas at the beginning of the third trimester of gestation. Insulin gene expression and islet cell biogenesis are dependent on several transcription factors (e.g., hepatocyte nuclear factor-4 [HNF-4α], HNF-1α, or HNF-1β; pancreatic and intestinal homeobox-1 [PDX1]; neuroD1) (Clinical Box 3.1).
The insulin gene encodes preproinsulin . Preproinsulin is converted to proinsulin by microsomal enzymes as the peptide enters the endoplasmic reticulum. Proinsulin is packaged in the Golgi apparatus into membrane-bound secretory granules. Proinsulin contains the AA sequence of insulin plus the 31-amino acid C (connecting) peptide . The proteases that leave proinsulin, prohormone (or proprotein) convertase-2 and -3, are packaged with proinsulin within the secretory vesicle. The mature hormone consists of two chains, an α chain and a β chain, connected by two disulfide bridges ( Fig. 3.3 ). Insulin is stored in secretory vesicles in zinc-bound crystals. Because the entire contents of the granule are released, equimolar amounts of insulin and C peptide are secreted, as are small amounts of proinsulin. C peptide has no known biologic activity, and proinsulin has about 7% to 8% of the biologic activity of insulin.
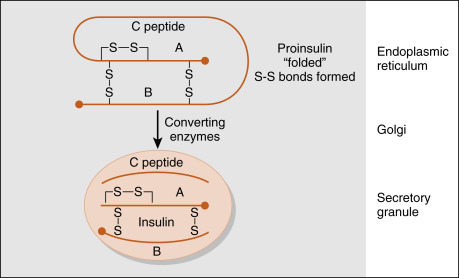
Measurements of C peptide in the blood are used to quantify endogenous insulin production in patients receiving exogenous insulin, which has been purified from C peptide. Insulin has about a 5-minute half-life and is cleared rapidly from the circulation by receptor-mediated endocytosis. It is degraded by lysosomal insulin degrading enzyme (IDE) in the liver, kidneys, and other tissues. Because insulin is secreted into the hepatic portal vein, almost one half of the insulin is degraded before leaving the liver. Recombinant human insulin and insulin analogs are now available, with different characteristics of onset and duration of action and peak activity.
Serum insulin levels normally begin to rise within 10 minutes after food ingestion and reach a peak in 30 to 45 minutes. The higher serum insulin level rapidly lowers blood glucose to baseline values. When insulin secretion is stimulated, insulin is released rapidly (within minutes), and this is called the early phase of insulin secretion. If the stimulus is maintained, insulin secretion falls within 10 minutes and then slowly rises over a period of about 1 hour. The second phase is referred to as the late phase of insulin release.
Glucose is the primary stimulus of insulin secretion ( Fig. 3.4 ). Glucose entry into β cells is facilitated by the GLUT2 transporter . Once glucose enters the β cell, it is phosphorylated to G6P by the low-affinity hexokinase, glucokinase (GK) . GK is referred to as the glucose sensor of the β cell because the rate of glucose entry is correlated to the rate of glucose phosphorylation, which, in turn, is directly related to insulin secretion. Heterozygous mutations in GK are one defect that leads to inadequate insulin release in patients with mature-onset diabetes of the young (MODY) . G6P is metabolized by β cells, increasing the intracellular ATP levels and closing an ATP-sensitive K + channel (see Fig. 3.4 ). This results in depolarization of the β-cell membrane, which opens voltage-gated Ca 2+ channels. Increased intracellular Ca 2+ levels activate exocytosis of secretory vesicles.
Heterozygous mutations in any one of the islet transcription factors, as well as glucokinase (see later), result in progressively inadequate production of insulin. This leads to MODY, which typically manifests in patients younger than 25 years of age.
- •
TYPE gene mutated (requires insulin or oral hypoglycemic)
- •
MODY 1 HNF-4α (yes)
- •
MODY 2 Glucokinase (no)
- •
MODY 3 HNF-1α (yes)
- •
MODY 4 IPF-1 (yes)
- •
MODY 5 HNF-1β (yes)
- •
MODY 6 Neuro D1 (yes)
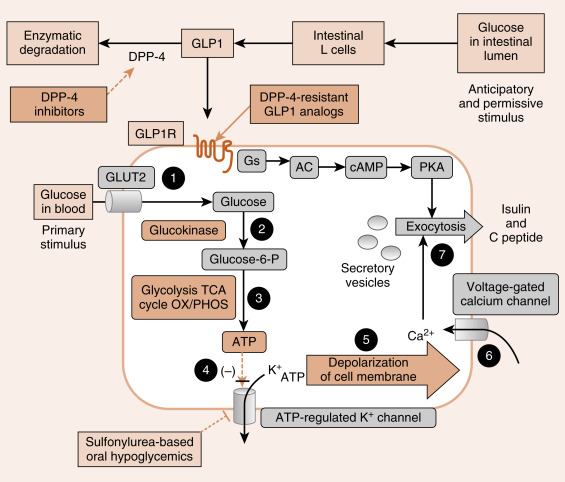
In addition to glucose, certain AAs (leucine) and vagal (parasympathetic) cholinergic innervation (i.e., in response to a meal) also stimulate insulin through increasing intracellular Ca 2+ levels. Long-chain FFAs also increase insulin secretion, although to a lesser extent than glucose and AAs.
As discussed in Chapter 2 , nutrient-dependent stimulation of insulin release is enhanced by the incretin hormones, glucagon-like peptide-1 (GLP-1) and gastric inhibitory peptide (GIP) and possibly other gastrointestinal (GI) hormones (e.g., cholecystokinin [CCK]). These act primarily by raising intracellular cyclic AMP (cAMP) within the β cell, which amplifies the intracellular effects of glucose on Ca 2+ (see Fig. 3.4 ). Intracellular cAMP acts both through phosphokinase A (PKA)-dependent and EPAC (exchange protein activated by cAMP)–dependent pathways (see Chapter 1 ) in β cells. Incretin hormones minimally increase insulin secretion in the absence of glucose.
Insulin secretion is inhibited by α 2 -adrenergic receptors , which are activated by epinephrine (from the adrenal medulla) and norepinephrine (from postganglionic sympathetic fibers). The α 2 -adrenergic receptors are coupled to a Gi-containing trimeric G-protein complex that inhibits adenylyl cyclase and decreases cAMP levels. Adrenergic inhibition of insulin serves to protect against hypoglycemia, especially during exercise. Although somatostatin from D cells inhibits both insulin and glucagon, its physiologic role in pancreatic islet function is unclear. Nevertheless, somatostatinomas are malignant tumors of the islets that produce high levels of somatostatin, which can lead to mild glucose intolerance.
The ATP-sensitive K + channel is a protein complex that contains an ATP-binding subunit called SUR1 . This subunit is also activated by sulfonylurea and meglitinide drugs , which are used as ingested drugs ( oral hypoglycemics ) to treat hyperglycemia in patients with partially impaired β-cell function (see Fig. 3.4 ). As these drugs act within the direct pathway for glucose-stimulated insulin secretion (GSIS) , they are associated with hypoglycemic episodes as a side effect. Rare mutations in the ATP-sensitive K + channel that keep it in the open conformation, thereby blocking glucose-induced insulin secretion, result in early-onset diabetes.
Analogs of GLP-1 (so-called “glutide” drugs ) are also used to enhance GSIS (see Fig. 3.4 ).
GLP-1 is rapidly degraded by the enzyme dipeptidyl dipeptidase-4 (DPP-4) . Inhibitors of DPP-4 (so-called “gliptin” drugs ) are also effective at enhancing GSIS. Both glutides and gliptins confer a significantly lower risk of hypoglycemia, as they only act in the presence of elevated glucose.
Insulin Receptor
The insulin receptor is a member of the receptor tyrosine kinase (RTK) family (see Chapter 1 ), which includes receptors for several other growth factors, such as IGFs, platelet-derived growth factor (PDGF), and epidermal growth factor (EGF). The insulin receptor is expressed on the cell membrane as a homodimer composed of α/β monomers ( Fig. 3.5 ). The α/β monomer is synthesized as one protein, which is then proteolytically cleaved, with the two fragments connected by a disulfide bond. The two α/β monomers are also held together by a disulfide bond between the α subunits. The α subunits are external to the cell membrane and contain the hormone-binding site. The β subunits span the membrane and contain tyrosine kinase on the cytosolic surface.
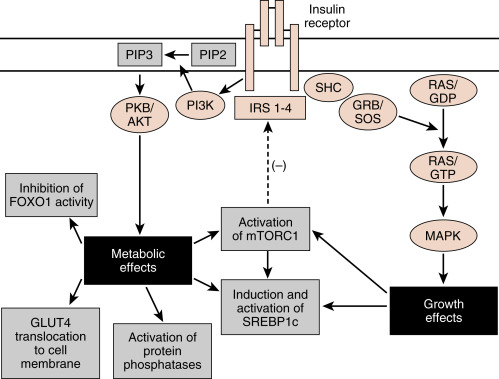
Insulin binding to the insulin receptor induces the β subunits to cross-phosphorylate each other on three tyrosine residues. These phosphotyrosine residues recruit two major classes of adaptor proteins: insulin-receptor substrate isoforms 1 to 4 (IRS1-4) and Shc protein (see Fig. 3.5 ). The IRS proteins are phosphorylated by the tyrosine kinase activity of the insulin receptor. The phosphotyrosine residues on IRS recruit PI3 kinase to the membrane, where it is phosphorylated and activated by the insulin receptor. PI3 kinase converts phosphoinositide-4,5-bisphosphate (PIP2) to phosphoinositide-3,4,5-triphosphate (PIP3). PIP3 recruits to the membrane and leads to the activation of a pleiotropic protein kinase, called Akt (see Fig. 3.5 ).
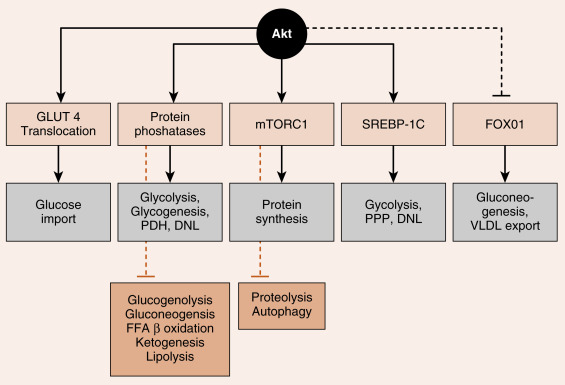
Akt regulates numerous enzymes and transcription factors that mediate the metabolic actions of insulin. Akt acts in five general ways that largely account for the metabolic effects of insulin ( Fig. 3.6 ):
- 1.
Phosphorylation of exocytosis components that induce the insertion of GLUT4 glucose transporters into the cell membranes of muscle and adipose tissue (see later). This action requires combined IRS/PI3K-dependent signaling and an additional adaptor protein-dependent pathway that activates a small G-protein pathway (not shown).
- 2.
Activation of protein phosphatases that, in turn, regulate metabolic enzymes through dephosphorylation. For example, dephosphorylation of liver pyruvate kinase activates the enzyme, whereas phosphorylation by cAMP/PKA signaling inactivates it.
- 3.
Activation of mammalian target of rapamycin complex-1 (mTORC1) (see later). mTORC1 is a kinase complex that promotes ribosomal RNA synthesis, ribosome assembly, and protein synthesis. mTORC1 also increases sterol-regulatory element–binding protein-1c (SREBP1c) activity.
- 4.
Direct induction of synthesis and activation of the lipogenic transcription factor, the SREBP1c. SREBP1c coordinately stimulates the expression of enzymes involved in glycolysis, lipogenesis, and the pentose phosphate pathway.
- 5.
Phosphorylation and inactivation of the hepatic transcription factor, FOXO1 .
- 6.
FOXO1 stimulates the expression of some gluconeogenic enzymes, as well as VLDL-related apoprotein B100.
The Shc protein is linked to the mitogen-activated protein kinase (MAPK) pathway (see Fig. 3.5 ), which mediates the growth and mitogenic actions of insulin (in conjunction with the activation of mTORC1).
The termination of insulin receptor signaling is a topic of high interest because these mechanisms potentially play a role in insulin resistance . Insulin induces the downregulation of its own receptor by receptor-mediated endocytosis . Additionally, several serine and threonine protein kinases are indirectly activated by insulin and by other molecules (such as inflammatory cytokines) and subsequently inactivate the insulin receptor or IRS proteins. These include mTORC1, which negatively feeds back on IRS proteins (see Fig. 3.5 ). Another serine threonine kinase that reduces activity or levels of the insulin receptor and IRS proteins is the suppressor of cytokine signaling (SOCS) family of proteins. In the presence of inflammation , proinflammatory cytokines activate SOCS kinase, which negatively feeds back on both the cytokine receptors and the insulin receptor. Thus inflammation can lead to insulin resistance. Finally, intracellular accumulation of TGs and other lipids increase cytosolic levels of lipid intermediates such as diacylglycerol (DAG) and ceramide . These intermediates lead to the activation of serine/threonine kinases such as protein kinase C (PKC) isoforms that phosphorylate and inhibit the insulin receptor and IRS proteins.
Glucagon
Glucagon is an important counterregulatory hormone that opposes insulin actions, and increases blood glucose levels through its effects on liver glucose output. Glucagon promotes the production of glucose through elevated hepatic glycogenolysis and gluconeogenesis and through decreased glycolysis and glycogen synthesis. Glucagon inhibits hepatic FFA synthesis ( de novo lipogenesis ) from glucose. Glucagon also maintains blood glucose indirectly through stimulation of ketogenesis , which provides an alternative energy source that leads to glucose sparing in many tissues.
Glucagon Structure, Synthesis, and Secretion
As discussed in Chapter 2 , glucagon is a member of the secretin gene family . Preproglucagon is proteolytically cleaved in the pancreatic islet α cell in a cell-specific manner to produce the 29-AA glucagon (see Fig. 2.9 ). Glucagon is highly conserved among mammals.
Like insulin, glucagon circulates in an unbound form and has a short half-life (about 6 minutes). The predominant site of glucagon degradation is the liver, which degrades as much as 80% of the circulating glucagon in one pass. Because glucagon (either from the pancreas or the gut) enters the hepatic portal vein and is carried to the liver before reaching the systemic circulation, a large portion of the hormone never reaches the systemic circulation. The liver is the primary target organ of glucagon, with lesser effects on adipose tissue. As discussed later, glucagon opposes the actions of insulin. Thus several factors that stimulate insulin inhibit glucagon. Indeed, it is the insulin-to-glucagon ratio that determines the net flow of hepatic metabolic pathways. A major stimulus for glucagon secretion is a drop in blood glucose, which is primarily an indirect effect via the removal of inhibition of glucagon secretion by insulin ( Fig. 3.7 ). Circulating catecholamines, which inhibit insulin secretion through α 2 -adrenergic receptors, stimulate glucagon secretion through β 2 -adrenergic receptors (see Fig. 3.7 ). Serum AAs also stimulate glucagon secretion. This means that a protein meal will increase postprandial levels of glucagon along with insulin, thereby protecting against hypoglycemia. In contrast, a carbohydrate-only meal stimulates insulin secretion and inhibits glucagon secretion.
Glucagon Receptor
The glucagon receptor is a 7-transmembrane receptor primarily linked to Gs-containing heterotrimeric G-protein complex (see Chapter 1 ). Consequently, glucagon increases intracellular cAMP levels in the liver. The increase leads to activation of protein kinase A (PKA). Glucagon stimulated PKA opposes the effects of insulin/Akt-activated protein phosphatases at several steps within hepatic and adipocyte metabolic pathways. The glucagon receptor is expressed by hepatocytes and adipocytes, but not by skeletal muscle.
Epinephrine and Norepinephrine
The other major counterregulatory factors are the catecholamines , epinephrine and norepinephrine . Epinephrine, and to a much lesser extent norepinephrine, are secreted by the adrenal medulla (see Chapter 7 ), whereas only norepinephrine is released from postganglionic sympathetic nerve endings. The direct metabolic actions of catecholamines are mediated primarily by β 2 – and β 3 -adrenergic receptors located on muscle, adipose, and liver. Like the glucagon receptor, β-adrenergic receptors are linked to a Gs signaling pathway that increases intracellular cAMP. Epinephrine also promotes glycogenolysis and gluconeogenesis through the α 1 -adrenergic receptor , which is coupled to the Gq/IP3/DAG signaling pathway.
Catecholamines are released from sympathetic nerve endings and the adrenal medulla in response to decreased glucose concentrations, stress or alarm, and exercise. Decreased glucose levels (i.e., hypoglycemia) are primarily sensed by hypothalamic neurons, which initiate a sympathetic response to release catecholamines.
Catecholamines circulate in the blood as free hormones, and both circulating and tissue catecholamines are rapidly enzymatically inactivated (see Chapter 7 ).
Intracellular Sensors and Regulators
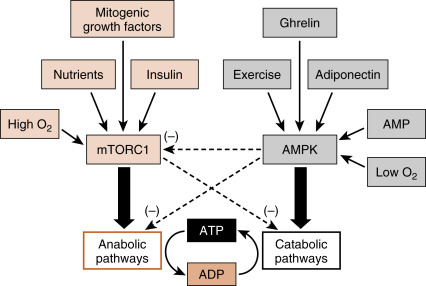
Importantly, all cells must balance energy needs associated with their differentiated function with other uses of carbon, especially the synthesis of macromolecules (lipids, nucleic acids, proteins) and the assembly of organelles that is associated with maintenance of cellular viability. There are several master regulatory nutrient and energy sensors that monitor intracellular nutrient levels and the relative amount of ATP to balance anabolic pathways with catabolic ones.
Two of these regulatory factors are mTORC1 and AMP-activated kinase (AMPK ; also a multimeric complex). In the presence of insulin and high levels of intracellular nutrients (AAs, glucose) that signal an abundance of carbons, mTORC1 promotes energy-consuming anabolic pathways, such as ribosomal RNA production and ribosome assembly, protein synthesis, and lipogenesis ( Fig. 3.8 ). Also, activation of mTORC1 inhibits the breakdown of intracellular macromolecules and organelles for energy and survival, a process termed autophagy . In contrast, a drain on energy supply associated with an elevation of AMP levels, or an indication of high energy use such as elevated intramuscular Ca 2+ levels and or reduced O 2 , activates AMPK (see Fig. 3.8 ). Hormones associated with a fasting state (ghrelin, adiponectin; discussed later) also activate AMPK. Active AMPK inhibits anabolic pathways (in part by inhibiting mTORC1 activity) and promotes energy-generating catabolic pathways, including glycolysis and β-oxidation of FFAs (see later). Although other important energy-sensing factors exist in cells, mTORC1 and AMPK have emerged as two centrally important factors involved in cellular energy balance, and the interaction of these two complexes with hormonal signaling is discussed later.
Metabolic Homeostasis: the Integrated Outcome of Hormonal and Substrate/Product Regulation of Metabolic Pathways
The hormonal regulation of the major metabolic pathways, with emphasis on key regulated enzymes and transporters, during the digestive phase compared with the fasting phase is presented in this section.
Energy Metabolism During the Digestive Phase
During the digestive phase, circulating nutrient levels increase. Following are some basic facts concerning the metabolism of glucose, FFAs and AAs during the digestive phase, with emphasis on hepatocytes, skeletal muscle and adipocytes.
Overview of Glucose Metabolism During the Digestive Phase
Glucose Transport Across the Cell Membrane
Glucose is a universal and normally abundant fuel. Glucose enters most cell types via facilitative, bidirectional transporters called GLUTs.
- 1.
GLUT1 is expressed in most cell types, including erythrocytes. GLUT1 deficiency is accompanied by severe neurologic disorders (e.g., epilepsy), emphasizing the importance of GLUT1 in the function of the blood-brain barrier and CNS.
- 2.
GLUT2 is a low affinity, high capacity transporter that is expressed in hepatocytes and pancreatic β cells . GLUT2 is paired with a low affinity, high V max hexokinase, GK, which can both act as a sensor of glucose levels, and allow for a high flux of glucose import.
- 3.
GLUT3 is expressed primarily in the CNS.
- 4.
GLUT4 is expressed primarily in skeletal muscle and adipocytes. During the fasting phase, GLUT4 is inactive because it resides within cytoplasmic “ glut storage vesicles .” In response to insulin signaling during the digestive phase, GLUT4-containing vesicles are inserted into and fuse with the cell membrane, thereby targeting the GLUT4 transporters to the cell membrane where they can actively import glucose. Because skeletal muscle (and in some individuals, adipocytes) represent an abundant cell type, insulin-dependent GLUT4 translocation and activation plays a central role in the removal of glucose from the circulation after a meal. Insulin resistance leads to deficient GLUT4 insertion into the cell membrane of skeletal myocytes and adipocytes, which contributes significantly to excessive blood glucose levels associated with glucose intolerance and type 2 diabetes mellitus ( T2DM ).
In addition to GLUT transporters, there also exist sodium/glucose-linked transporters, SGLT1 and SGLT2 , primarily found in the brush borders of the small intestine and proximal convoluted tubule , respectively. Sodium is actively transported out of the cell through the basolateral membrane by an Na + /K + -ATPase . This allows sodium to be transported through SGLTs into the cell down a concentration gradient. SGLTs are symporters, in that glucose is transported into cells along with sodium. Glucose is then exported through the basolateral membrane through GLUT2 transporters, and then enters capillary beds through GLUT1 transporters in endothelial cells. SGLT2 is highly expressed in the proximal (S1) segment of the proximal convoluted tubule, and plays a major role in the recapture of sodium and glucose that exit the blood at the glomerulus. Normally, 100% of filtered glucose is recaptured by the proximal convoluted tubule and returned to the circulation. “Gliflozin” drugs inhibit SGLT2 and thus renal reabsorption of glucose, and are currently being used to treat T2DM .
Utilization of Glucose
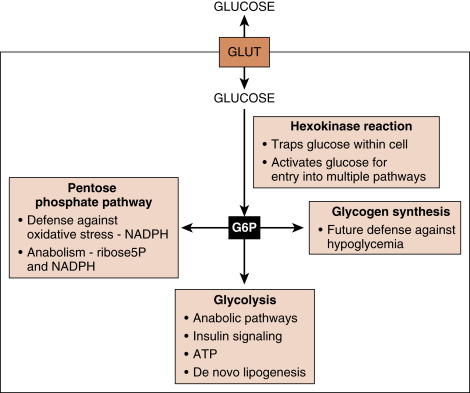
Glucose is rapidly phosphorylated to G6P by the family of hexokinases ( Fig. 3.9 ). This reaction traps glucose within the cell, in that G6P cannot exit through bidirectional GLUT transporters. The hexokinase reaction also “activates” glucose, so that as G6P, the carbons within glucose can potentially enter several pathways (see Fig. 3.9 ):
- 1.
Glycogen synthesis in hepatocytes and skeletal muscle. Glycogen is a storage form that can be accessed during fasting and exercise.
- 2.
Pentose phosphate pathway (PPP). The PPP generates riboses that are used for nucleotide and nucleic acid synthesis, as well as NADPH , which is a cofactor for antioxidant defense pathways and anabolic pathways.
- 3.
Glycolysis:
- a.
Glycolysis converts G6P to pyruvate and generates a net of two molecules of ATP/G6P. Glycolysis occurs in the cytoplasm and does not require O 2 , so that erythrocytes can use glucose to generate ATP, and convert pyruvate to lactate , which is exported.
- b.
Side reactions of glycolysis contribute to several pathways, including those involved in protein glycosylation, AA synthesis, single carbon metabolism that contributes to folate-dependent synthesis of nucleic acids, and glycerol-3-phosphate that is used for TG synthesis.
- a.
- 4.
Mitochondrial utilization of pyruvate . Most normal cells further metabolize pyruvate through mitochondrial respiration , which requires O 2 . This involves conversion of pyruvate to acetyl CoA by pyruvate dehydrogenase (PDH) complex , complete oxidation of acetyl CoA to CO 2 through the TCA cycle , and oxidation of the TCA cycle-generated NADH and FADH 2 through the electron transport system and ATP synthase to generate ATP.
- 5.
De novo lipogenesis— this is discussed later in the context of hepatocyte metabolism during the digestive phase.
Glucose is produced and exported by the liver through glycogenolysis and gluconeogenesis , collectively referred to as hepatic glucose production. Insulin has direct inhibitory actions on hepatic glucose production during the digestive phase. It must be stressed that glucose is a reactive molecule and prolonged, inappropriately elevated levels of circulating and, consequently, intracellular glucose leads to the nonenzymatic glycation of proteins and lipids. Glycation of these molecules leads to aberrant processing, folding, secretion (e.g., of extracellular matrix proteins), and function. Glucose also contributes to the osmolarity of fluids, so that as excessive glucose is cleared by the kidney, it drags water with it, causing potentially dangerous dehydration . Therefore it is important that blood glucose levels remain below a critical threshold (e.g., fasting blood glucose should be below 100 mg/dL). Chronic elevation of glucose, due to the inability of certain tissues/organs to import and metabolize glucose, and/or due to inappropriate hepatic glucose production, is defined as glucose intolerance (fasting blood glucose between 100 to 125 mg/100 dL); or diabetes mellitus (DM) (fasting blood glucose higher than 125 mg/100 dL). As discussed later, DM imposes a broad range of stresses on cells that ultimately are manifested by the compromised function or failure of specific organs. Thus it is important to understand that insulin imposes important negative actions on metabolism that contribute to the maintenance of blood glucose levels below the upper normal limit (i.e., below 100 mg/dL for an overnight fasting glucose).
Overview of Metabolism of Selected Amino Acids During the Digestive Phase
The flow of AAs into hepatocytes during the digestive period is coupled to deamination and entry of ammonia into the urea cycle, and the conversion of deaminated carbon skeletons of AAs into pyruvate, acetyl CoA, or intermediates of the TCA cycle ( Fig. 3.10 ). AAs are also used in several anabolic pathways, including of course protein synthesis, but also including glutathione synthesis, nucleotide synthesis, and glucosamine synthesis during the digestive phase. Insulin both promotes the synthesis of many macromolecules (e.g., protein synthesis), as well as inhibiting their degradation (e.g., proteosomal degradation of proteins).
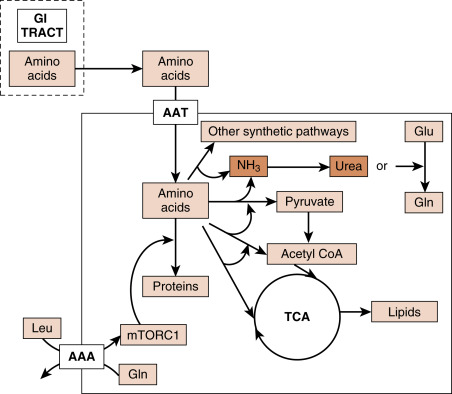
AAs that are not metabolized by the liver enter the general circulation for use by other cells. The most abundant circulating AA is glutamine (Gln), due to its abundance in the diet and to its generation from the transfer of amino groups to glutamate (Glu) in most cells. Gln is required for the hexosamine pathway that is involved in protein glycosylation, nucleotide biosynthesis, and can be exchanged for leucine (Leu) through several cell membrane antiporters. Leu, in turn, is a strong activator of mTORC1, which promotes RNA, protein and lipid synthesis (see Fig. 3.10 ). Leu also stimulates insulin secretion in β cells. Gln can also be converted to Glu, which can be used to synthesize several other AAs, protein, as well as the antioxidant molecule, glutathione. Glu can also be converted to the TCA cycle intermediate, α-ketoglutarate (also designated 2-oxoglutarate). This is especially important in the hepatocyte during de novo lipogenesis, in that α-ketoglutarate replaces carbons (“anaplerosis”) that exit the TCA cycle as citrate (“cataplerosis”). Thus Gln and Glu contribute to de novo lipogenesis, as well as ATP generation. Other AAs can be converted to pyruvate or TCA cycle intermediates and also contribute to de novo lipogenesis and ATP production.
Overview of Long-Chain Free Fatty Acid Metabolism During the Digestive Phase
Long chain FFAs (designated herein simply as FFAs ), with 13 to 21 carbons, are common in the diet. These lipids are absorbed by the intestinal enterocytes as either FFAs or as 2-monoglyceride. Within the enterocytes, FFAs are re-esterified to the 2-monoglyceride to form TG. TG, in turn, is assembled with the apoprotein B48 and other lipids to form nascent chylomicrons . Chylomicrons are too large to enter capillaries, and instead enter lymphatics that convey them to the thoracic duct that empties directly into the circulation near the left venous angle. Thus unlike glucose and AAs, chylomicrons are not directly conveyed to hepatocytes via the hepatic portal vein, but enter the systemic circulation close to the heart (see Fig. 3.1A ).
Within the circulation, chylomicrons mature by having other apoproteins transferred to them from high-density lipoprotein (HDL) particles. As chylomicrons enter the capillary beds within adipose tissue, they undergo lipolysis that is catalyzed by lipoprotein lipase (LPL) . In response to high insulin during the digestive phase, LPL is synthesized within adipocytes, secreted, and ultimately becomes noncovalently attached to the luminal membrane of the endothelial cells that line the capillary beds. LPL-mediated digestion releases FFAs from chylomicron-associated TGs (discussed later). The FFAs are transported across the endothelial cells and into the adipocytes. As stated previously, adipocytes convert a significant fraction of glucose to glycerol-3-phosphate, which is used as the backbone for reesterification of the imported FFAs for storage as TG within the adipocyte.
Several intracellular lipases can reverse this process, potentially releasing FFAs into the blood through an inappropriate futile cycle during the digestive phase. FFAs would compete for glucose utilization, thereby promoting glucose intolerance. A key hormone-regulated intracellular lipase is called hormone-sensitive lipase (HSL) . During the digestive phase, high insulin levels directly inhibit HSL activity and prevent the lipolysis of newly synthesized TG.
TG is stored as cytoplasmic lipid droplets (CLDs) within adipocytes. These CLDs are coated primarily by the perilipin isoform, PLIN1. Perilipins interact with lipases and lipase-activating proteins that stabilize CLDs during the digestive phase, but reconfigure CLDs in the presence of PKA signaling from catecholamines and glucagon to allow for lipolysis and release of FFAs and glycerol.
The partially digested chylomicrons become chylomicron remnants as they move through adipose capillary beds. Chylomicron remnants eventually bind to apoprotein E–related receptors on hepatocytes and undergo receptor-mediated endocytosis and digestion within lysosomes. The residual FFAs that are released from the remnants are reesterified to TGs, and thus contribute to the total intrahepatic TG.
Hepatic Metabolism During The Digestive Phase
Anatomic Considerations
The liver is anatomically and functionally situated between the GI tract and the heart. The main veins that drain the GI tract of its ingested nutrients all converge to form the hepatic portal vein. A “portal vein” is defined as a vein that conveys the contents of a capillary bed (or many convergent capillary beds) to a second set of capillary beds before reaching the heart. In the case of the hepatic portal vein, it receives the venous drainage of the GI tract and then ends at the hepatic sinusoids (a sinusoid is a discontinuous capillary). This means that the liver is the first organ to receive the ingested carbohydrates and AAs before they reach the general circulation (as stated previously, FFAs and TGs initially bypass the liver because they enter lymphatics as chylomicron particles). The liver is also the first organ to respond to hormones and cytokines from the pancreas and intraabdominal adipose tissue.
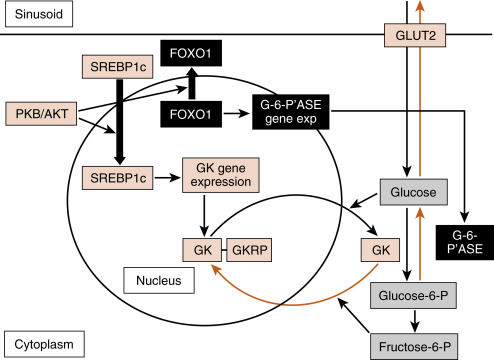
Hormonal Regulation of Key Reactions in the Liver During the Digestive Phase
Intracellular Transport and Trapping of Monosaccharides
Ingested monosaccharides ( glucose, fructose, and galactose ) reach the liver sinusoids and flow down their concentration gradient into hepatocytes through the bidirectional, facilitative transporter, GLUT2 ( Fig. 3.11 ). GLUT2 is a low-affinity, high-capacity facilitative transporter well-suited to moving high amounts of absorbed glucose during and soon after a meal. GLUT2 expression or localization at the hepatocyte cell membrane is largely not regulated.