Abstract
The success of pregnancy depends on (1) fertilization and successful implantation of the developing embryo into the endometrium; (2) development and function of the placenta; (3) adaptation of maternal physiology to accept the fetal allograft and satisfy its nutritional, metabolic, and physical demands; (4) appropriate growth and functional development of key organ and homeostatic systems in the fetus; and (5) proper timing of birth so that it occurs when the fetus is mature enough to survive outside the uterus. This chapter examines the complex array of hormonal interactions between the fetus, placenta, and mother that controls these processes.
Keywords
Pregnancy, hormones, placenta, implantation, parturition
Introduction
The birth of a healthy baby is dependent on an ordered sequence of biological events during pregnancy. Important among these are:
- •
Successful implantation of the developing embryo
- •
Adaptation of maternal physiology to accept the fetal allograft and satisfy its nutritional, metabolic, and physical demands
- •
Appropriate growth and functional development of key organ and homeostatic control systems in the fetus in preparation for life outside the uterus
- •
Proper timing of birth so that it occurs when the fetus is mature enough to survive outside the uterus
The hormonal interactions between the fetus, placenta, and mother that control these processes are discussed in this chapter.
The chapter begins with a brief review of the hormonal events that control endometrial growth and function during the menstrual cycle, so that it becomes receptive to blastocyst implantation and the establishment of pregnancy. The processes of implantation and placental development and the mechanism by which the fetal allograft overcomes attack by the maternal immune system are also discussed.
Once pregnancy is established, the fetus, placenta, and mother initiate and maintain communication via the endocrine system. The endocrine milieu of human pregnancy is dominated by placental hormones, the major function of which is to modify maternal physiology to satisfy the nutritional, metabolic, and physical demands of the growing fetus. In this context, the physiologic role of the principal placental hormones is discussed.
The fetal neuroendocrine system must become functionally competent by term so that the neonate can maintain homeostasis after birth. These systems are also critical for the fetus’ future health. The fetal programming hypothesis posits that adaptation of key fetal neuroendocrine systems to the intrauterine environment affects their function during postnatal life, and in some cases, this adaptation can be the origin of adult diseases such as metabolic syndrome and obesity. Development of fetal neuroendocrine systems and the influence of the fetal-maternal environment are therefore discussed separately.
Finally, the endocrine interactions between the fetus, placenta, and mother that culminate in the induction of parturition are reviewed. The appropriate timing of birth, so that it occurs when the fetus is sufficiently mature to survive as a newborn, is a major determinant of postnatal survival in all viviparous species. Current understanding of the hormonal interactions that control the process and timing of human parturition is described.
Establishment of Pregnancy
- ◆
The establishment of pregnancy involves complex endocrine and paracrine signaling between the embryo and the mother to keep the uterus in a state conducive for implantation, shield the embryo from attack by the maternal immune system, and remodel the endometrium to facilitate implantation and placentation.
The first step in the establishment of pregnancy is implantation of the blastocyst into the endometrium. The goal of the embryo is to establish a conduit through which it can access a supply of nutrients from the mother. The process of embryo apposition, adhesion, and invasion into the endometrium involves a complex bidirectional hormonal dialogue between the blastocyst and the endometrium, such that embryo viability and endometrial receptivity are synchronized to allow for successful implantation and the establishment of pregnancy. As the embryo is semiallogeneic with respect to the mother, its invasion into the endometrium represents a major breach of the maternal immune system. The hormonal dialogue between the embryo and the mother at the implantation site is critical in preventing attack by the maternal immune system.
Endometrial Receptivity
During the menstrual cycle, the ovarian steroid hormones, estradiol and progesterone, induce structural and functional changes in the endometrium essential for implantation and the establishment of pregnancy (see Chapter 9 ). During the luteal phase and under the influence primarily of progesterone, the proliferative endometrium is converted to a secretory phenotype. It becomes thick (5 to 6 mm), spongy, and highly vascularized. Secretions from the glandular epithelium establish an intrauterine environment that favors embryo survival, and the epithelial cells produce a variety of chemokines, growth factors, and cell adhesion molecules (CAMs) that attract the blastocyst to specific docking sites for implantation. Increased vascularization and the development of spiral arterioles within the endometrial stroma provide an optimal substrate for invasion and placentation.
Receptivity of the endometrium to blastocyst adhesion and implantation has temporal and spatial characteristics. In humans, implantation between days 21 and 24 of the menstrual cycle is associated with a high (~85%) success rate for continuing pregnancy, whereas implantation after day 25 has a low success rate (~11%). The receptive phase of endometrial development is termed the implantation window , opening 5 to 6 days after ovulation and closing 3 to 4 days later. During the implantation window, the endometrial epithelium develops domelike structures known as pinopodes, which correlate with implantation sites. The tips of the pinopodes express chemokines and CAMs that attract the embryo and appear to be the preferred site for embryo adherence. Interestingly, areas between the pinopodes produce a repellent molecule, MUC-1, that prevents embryo adhesion. Analysis of the human endometrial transcriptome during the implantation window has revealed numerous genes that are up- and downregulated in comparison with late proliferative phase endometrium, demonstrating the remarkable complexity of molecular interactions underlying endometrial differentiation.
Implantation
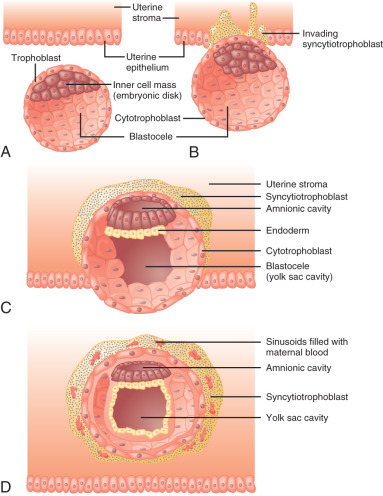
After fertilization, the embryo undergoes an intrinsic developmental program. The sequence of cell divisions and differentiation is not dependent on the hormonal milieu of the fallopian tube or the uterus, as fertilization and early embryonic development occur successfully in vitro. By the third to fourth day after fertilization, the embryo comprises a solid ball of cells encapsulated by the translucent remnant of the zona pellucida. At about the fifth day after fertilization, a fluid-filled cavity, the blastocele, forms within the embryo. At this stage the embryo is referred to as a blastocyst . The outer layer of blastocyst cells adjacent to the zona pellucida is known as the trophectoderm . These cells eventually give rise to the placenta and chorion. The fetus, amnion, and mesenchymal and vascular components of the placenta are derived from the inner cell mass, a group of cells lying under the trophectoderm at one end of the blastocele.
Although there is considerable variation in the process of implantation between eutherian mammals, the end result is the same: the blastocyst becomes fixed in position and forms a physical and functional contact with the uterus.
In humans, the blastocyst enters the uterus at around the fifth day after fertilization and floats freely for 2 to 3 days. By this stage, the blastocyst escapes the confines of the zona pellucida (i.e., it hatches) and is in an optimal condition to implant. The developing blastocyst interacts in a paracrine manner with the endometrial epithelium by producing multiple cytokines, chemokines, and CAMs that facilitate adherence.
Almost immediately after fertilization, hormonal signals from the conceptus are transmitted to the mother. During its journey through the fallopian tube, the cumulus cells surrounding the embryo produce progesterone and estrogen. These steroids are thought to act locally to modulate tubal motility to facilitate transport of the conceptus toward the uterine cavity. Studies of the secreted proteins (the secretome) produced by the preimplantation embryo show that the profile of secreted proteins changes every 24 hours and with the different embryonic stages. Thus a specifically orchestrated sequence of secretions from the embryo plays a key role in the paracrine interaction between the embryo and epithelial cells of the endometrium. Any delay in embryo development or in its transit through the oviduct disrupts the synchrony between blastocyst development and endometrial receptivity and increases the likelihood of reproductive failure. Thus development of the “seed” and conditioning of the “soil” are synchronized. Given this delicate balance, it is not surprising that the highest rate of embryo loss occurs during the periimplantation period.
The embryo implants in a polarized fashion. After hatching from the zona pellucida, trophoblast cells overlying the embryonic pole are the first to interact with the uterine epithelium ( Fig. 11.1 ). Those cells dock with specific sites in the epithelial lining of the endometrium, most likely at the pinopodes. The trophoblast cells proliferate and secrete proteases that degrade the extracellular matrix between endometrial cells, forming a path for the blastocyst to enter the uterine stroma. At this stage the trophoblast cells are referred to as cytotrophoblasts and differentiate along two distinct paths: villous and extravillous. The extravillous cytotrophoblasts become highly invasive and form columns penetrating the basal membrane beneath the decidual cells and into the myometrium. Eventually the entire embryo is embedded in the uterine stroma and anchored by villous cytotrophoblasts. During this time, some villous cytotrophoblasts fuse their plasma membranes to become a single multinucleated cell known as the syncytiotrophoblast, which becomes the outer layer of the functional floating villi in direct contact with maternal blood.
As with adhesion, implantation also involves a complex paracrine dialogue between the embryo and the endometrium-decidua. Many of the molecules (produced by the endometrial cells or the blastocyst) involved in the implantation process have been identified. These include:
- •
Leukemia inhibiting factor
- •
Interleukin-11
- •
Heparin-binding epidermal growth factor (EGF)–like growth factor
- •
Prostacyclin
- •
Prostaglandin E 2
- •
Peroxisome-proliferator-activator receptor
- •
HOXA10 and HOXA11
- •
Metalloproteinase-2 and -9
- •
Basic and acidic fibroblast growth factor (FGF)
- •
Bone morphogenetic proteins (BMPs) and the BMP antagonist Noggin
- •
WNT and Indian hedgehog signaling molecules
Decidualization of the Endometrium
Late in the luteal phase, the endometrial stroma undergoes a series of morphologic and functional changes collectively referred to as decidualization. The process of decidualization is dependent on progesterone and occurs independently of conception and implantation. It begins around blood vessels during the midsecretory phase and progressively encompasses the entire endometrial stroma during the latter part of the secretory phase. The fully decidualized endometrial stroma is composed of large polyhedral cells, containing high levels of glycogen and lipids, that secrete a tough pericellular capsule composed of collagen, laminin, fibronectin, and heparan sulfate proteoglycan. Bone marrow-derived immune cells, primarily large granular lymphocytes (LGLs), also accumulate, and extensive cell-to-cell contacts between the lymphocytes and the stromal cells form, especially in the vicinity of blood vessels.
The decidualized endometrium is hostile to implantation. Therefore the blastocyst has a limited window of opportunity to establish a successful implantation before the decidual barrier is established. Attachment of the blastocyst to the uterine epithelium accelerates the decidualization process, further limiting the window of opportunity for implantation. In an infertile cycle, menstruation occurs to eliminate the nonreceptive decidualized endometrium. The endometrium is renewed to a receptive state in the subsequent cycle. The term decidua is highly appropriate, because this tissue is cast off at menstruation, miscarriage, and delivery.
The decidua is thought to form a critical barrier between the mother and the conceptus and represents an attempt by the mother to restrict blastocyst invasion. The large component of immune cells (mainly LGLs) within the decidual tissue may also restrict the passage of fetal antigens to the maternal compartment. Interestingly, decidualization of the endometrium appears limited to species with interstitial implantation, in which the blastocyst is completely embedded in the endometrial stroma, further supporting the argument that it limits the invasiveness of the blastocyst during implantation.
Once pregnancy is established, the decidua can be divided into three types, depending upon anatomic location: (1) the decidua basalis, which underlies the site of implantation and forms the maternal component of the placenta; (2) the decidua capsularis, which overlies the gestational sac (this portion disappears in the later stages of pregnancy); and (3) the decidua vera, which lines the remainder of the uterine cavity and becomes intimately approximated to the chorion.
The decidua of pregnancy, in association with the fetal membranes, is considered to be an endocrine organ. Hormones produced by the decidua can act on the adjacent tissue (chorion and myometrium) or communicate with the fetus via the amniotic fluid. The decidua produces prolactin, relaxin, and prostaglandins (PGs) that are thought to be involved in the process of parturition (discussed later in the chapter). Its close proximity to the myometrium is ideal in this regard.
Placentation
Early in the implantation process, some cytotrophoblast cells aggregate and form migrating columns that penetrate into the inner third of the myometrium. The invading cytotrophoblasts target maternal blood vessels and, via interstitial and endovascular routes, completely surround and plug spiral arterioles. They then displace maternal endothelial cells and musculoelastic tissue and effectively create a low-resistance arteriolar system by increasing vessel diameter. Blood flowing from those vessels fills spaces, known as lacunae, between the invading columns of cytotrophoblast cells, and leave through venous connections. Eventually the syncytiotrophoblasts come into contact with maternal blood and form chorionic villi that are bathed in maternal blood within the lacunae.
According to Poiseuille’s law of fluid dynamics, flow through a cylinder is proportional to the radius multiplied to the fourth power. The average radius of arterioles in the pregnant human uterus is around 10-fold greater than in the nonpregnant state. Such a change would increase flow 10,000-fold, which is ample to serve the future requirements of the growing fetus. Cytotrophoblasts within the spiral arterioles not only facilitate this large diversion of maternal blood flow to the conceptus, but also prevent maternal vasomotor control. Through this process, the supply of resources to the conceptus is optimized, and the capacity for maternal restriction of uterine blood flow is subverted.
Placentation across all eutherian mammals is characterized by high angiogenic activity and blood vessel growth. This is particularly the case for the site of placental attachment. The human placenta produces several known angiogenic factors, including platelet-derived endothelial cell growth factor, vascular endothelial growth factor (VEGF), and angiopoietin-1 and angiopoietin-2. In addition, two potent inhibitors of angiogenesis have been isolated from mouse placenta. The specific interplay between angiogenic factors and their inhibitors in the placenta is not well understood. However, it is possible that the presence of antiangiogenic factors within the placenta prevent maternal endothelial cells from resealing the ends of spiral arterioles that have been occupied by trophoblasts. In addition, antiangiogenic factors may prevent the overgrowth of maternal and fetal vessels, thereby preventing maternal blood vessels from entering the fetal compartment and fetal vessels from extending beyond the uterus.
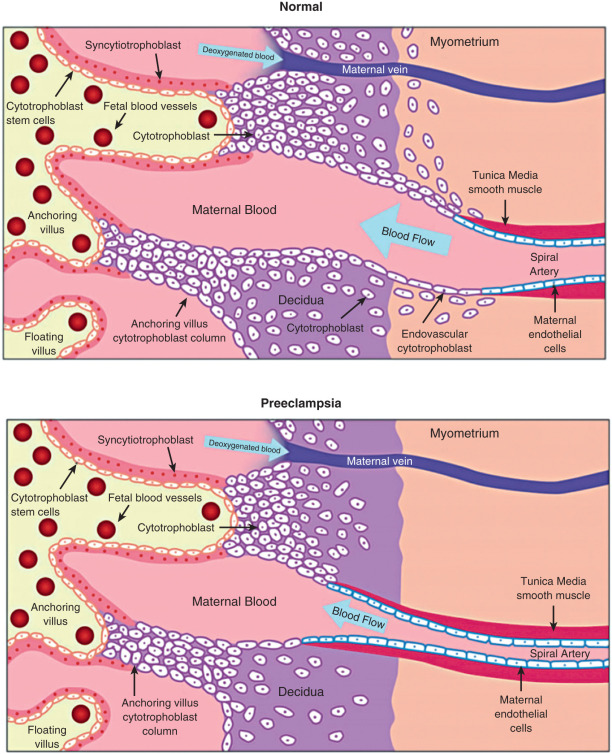
The angiogenic events occurring during implantation and placentation likely are critical factors in the etiology of hemodynamic disorders in pregnancy. The extent and depth of the placental incursion appear to be the sum of the intrinsic proinvasive characteristics of trophoblasts and the physical and biochemical barriers mounted by the maternal tissues. Imbalances in this equation can lead to failed implantation and pathologic conditions. The arteriole remodeling process is complex and requires the cytotrophoblast cells to express specific adhesion molecules and adopt an endothelial phenotype. If this process is impaired, the depth of invasion and remodeling may be limited, leading to intermittent perfusion and placental ischemia ( Fig. 11.2 ). Abnormal spiral artery remodeling is associated with fetal growth restriction and hypertensive disorders of pregnancy. In preeclampsia, cytotrophoblast invasion is restricted to the superficial decidual segments, leaving the myometrial spiral arterioles undisturbed and still responsive to maternal vasomotor influences. To compensate, especially during the later stages of pregnancy when nutritional requirements of the fetus increase, the placenta secretes factors into the maternal circulation that increase maternal systemic blood pressure.
Because cytotrophoblasts are genetically distinct from maternal tissue, implantation represents an extraordinary breach of maternal immune defenses. The mother must distinguish between the fetal allograft as being immunogenically foreign or self, and/or the placental trophoblasts must protect themselves from immune destruction by activated maternal immune cells. The decidua is composed mainly of a uterus-specific subset of LGLs with natural killer (NK) cell phenotype. NK cells preferentially attack targets lacking polymorphic Class I or Class II human leukocyte antigens (HLAs). Trophoblasts have a unique repertoire of HLAs and do not express polymorphic HLA-A, HLA-B, and HLA-DR, and therefore are highly susceptible to attack by decidual NK cells. However, differentiated cytotrophoblasts that lie at the maternal-fetal interface produce a unique Class I molecule known as HLA-G. Because of its reduced polymorphism, HLA-G is less immunogenic and could prevent immunologic recognition by NK cells. In addition, to repress the maternal immune system, trophoblast cells express multiple factors, including Fas ligand, which induces apoptosis in immune cells that carry the Fas receptor, early pregnancy factor, and progesterone-induced blocking factor that block lytic NK cell activity and several antiinflammatory cytokines (e.g., TGF-β, IL-10, IL-4). Thus the fetal allograft may escape immune attack by presenting the unique HLA-G camouflage or by actively subverting immune recognition via multiple cytokine-based mechanisms.
The functional unit of the human placenta is the chorionic villus, which is formed by a sprouting of syncytiotrophoblasts that project into a pool of maternal blood surrounding the blastocyst. The fingerlike chorionic villi have a central core of loose connective tissue with an extensive capillary network linking it with the fetal circulation. Surrounding the core are the outer syncytiotrophoblast and inner cytotrophoblast cells that form the functional barrier between the maternal and fetal circulations. This arrangement is referred to as hemochorial placentation , since the maternal blood is in direct contact with the fetal chorion (syncytiotrophoblast) layer.
Endocrine Placenta
- ◆
The placenta produces a plethora of hormones that modulate maternal physiology to maintain pregnancy and provide the fetus with resources needed for growth and development.
Placental hormones dominate the endocrine milieu of human pregnancy. This remarkable organ produces a plethora of hormones that it secretes in large quantities primarily into the maternal circulation ( Table 11.1 ). The hemochorial arrangement of the human placenta is ideal for this purpose. Cytotrophoblast and syncytiotrophoblast cells of the placenta have direct access to the maternal circulation. In contrast, the syncytiotrophoblast layer prevents most maternal hormones from entering the fetal compartment. Consequently, the fetal-placental endocrine system generally develops and functions independently of that of the mother.
| Neuropeptides | Pituitary-like Hormones | Adipokine | Growth Factors | Steroid Hormones | Monoamines and Adrenal-like Peptides |
|---|---|---|---|---|---|
|
|
|
|
|
|
Most placental hormones are counterparts to those produced in the nonpregnant adult, and therefore bind to cognate receptors on maternal cells. In this context, placental hormones may be regarded as allocrine factors—that is, hormones produced by one organism (the fetus) to act on the receptors of another (the mother). Remarkably, the placenta produces and responds to functional cohorts of hypothalamic and pituitary hormones and functions as a secondary neuroendocrine control center. This “extra” brain in many instances overrides the maternal system to adjust maternal physiology, usually in favor of supporting the needs of the fetus. The role of some of the principal placental hormones in the endocrine control of human pregnancy—especially the maternal adaptations to pregnancy—is discussed below.
Placental Hypothalamic-Pituitary-Like Hormones
Chorionic Gonadotropin
One of the first hormonal signals from the embryo to the mother prevents mensuration and sustains an endometrial lining that is receptive for implantation. This is achieved by the secretion of a gonadotropin-like glycoprotein hormone, chorionic gonadotropin (CG, designated hCG in humans), produced by the trophoblastic cells of the early embryo. hCG acts on the corpus luteum (CL) to prevent luteolysis and maintain steroidogenic function. This causes progesterone levels to be sustained. Progesterone is required to maintain the endometrium in the secretory state. In nonconceptive cycles, the CL usually regresses at about the second week after ovulation, and the subsequent decline in progesterone leads to menstruation. For pregnancy to be established, CL regression and the associated decrease in systemic progesterone must be prevented. Thus one of the first endocrine interactions between the conceptus and the mother involves signaling by the early embryo to promote intrauterine conditions that will allow implantation and the establishment of pregnancy. This is referred to as the maternal recognition of pregnancy and involves extending the functional lifespan of the CL.
As with other glycoprotein hormones, such as luteinizing hormone (LH) and follicle stimulating hormone (FSH), hCG is a heterodimer composed of α- and β-subunits. The α-subunits of hCG, LH, and FSH are identical, whereas the β-subunits differ, conferring specificity to each hormone. hCG is biologically and immunologically similar to pituitary LH. The α-subunit of hCG is produced by cytotrophoblast cells, especially as they differentiate during implantation. The β-subunit of hCG is primarily produced by the syncytiotrophoblast, but can also be detected in mature cytotrophoblast cells just before they fuse to form the syncytiotrophoblast. The syncytiotrophoblast produces both subunits and is the principal source of functional hCG.
Multiple variants of hCG have been identified in the human placenta. In addition to native hCG produced by the syncytiotrophoblast, extravillous invasive cytotrophoblasts produce hyperglycosylated hCG. The three hCG variants have distinct physiologic functions. Like LH, native hCG is a potent luteotropin, and as such, it stimulates progesterone secretion by the CL. The hyperglycosylated hCG and the monomeric β-subunit are thought to exert primarily extragonadal actions and are associated with malignancies. The actions of all hCG variants are mediated through interaction with the LH receptor.
In an evolutionary context, production of hCG may provide a basis for selection, as pregnancy will not occur if the embryo cannot gain control of the CL. The capacity of the embryo to produce large amounts of hCG may represent a selective test of the embryo’s endocrine competence. The maternal level of resistance to the embryo’s efforts to control the CL would select embryos with more robust endocrine function.
During the first 5 to 7 weeks of pregnancy, the majority of progesterone is produced by the CL in response to hCG. Consequently, the ovaries are obligate organs for pregnancy maintenance during this time, and abortion rapidly ensues if they are removed. However, after weeks 6 to 7 of pregnancy, the placenta begins producing progesterone, and at around the same time, progesterone production by the CL decreases. Thereafter the placenta supplies the bulk of progesterone for the remainder of pregnancy. The transition in the source of progesterone is referred to as the luteal-placental shift .
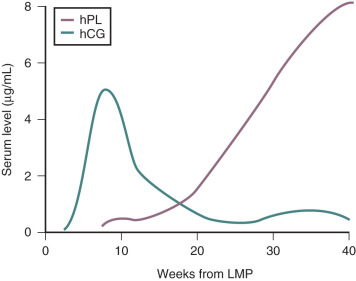
In normal pregnancies, hCG is detectable 9 to 11 days after the midcycle LH peak, which is around 8 days after ovulation and only 1 day after implantation. Therefore pregnancy can be detected before the first missed menstrual period. This has clinical utility when it is important to determine the presence of pregnancy at an early stage. In early pregnancy, there is an approximate doubling of circulating hCG levels every 2 to 3 days, and concentrations of hCG rise to peak values by 60 to 90 days of gestation. Thereafter hCG levels decrease to a plateau that is maintained during the remainder of the pregnancy ( Fig. 11.3 ). Assayable LH and FSH levels in the maternal blood are virtually undetectable throughout pregnancy.
The actions of hCG may not be limited to maintaining progesterone production by the CL. Much of the increased thyroid activity that occurs in pregnancy has been attributed to hCG, which binds specifically to thyroid cell membranes and displaces thyroid-stimulating hormone (TSH). hCG also influences the development and function of the fetal adrenals and testes. In addition, hCG may have actions on the maternal reproductive tract, including the decidual response, relaxin production by the CL, and relaxation of uterine smooth muscle. hCG receptors have been detected in fetal membranes and in vitro hCG inhibits spontaneous contractility in strips of human myometrium.
Production of hCG by the invading blastocyst may contribute in a paracrine manner to the implantation process. In vitro and in vivo studies in nonhuman primates demonstrate that hCG promotes decidualization of the endometrial stroma. The hormone appears to have marked effects on endometrial physiology at the implantation site before its levels are detectable in the circulation. Studies suggest that hCG produced by the blastocyst prolongs the window of implantation by inhibiting endometrial insulin-like growth factor binding protein-1 (IGFBP-1) production, augmenting angiogenesis at the implantation site by increasing VEGF expression, modulating local cytokine and chemokine expression, augmenting local protease activity, and, via an autocrine effect on the trophoblasts themselves, promoting differentiation and augmenting invasive potential.
Placental trophoblasts may have an autoregulatory mechanism for hCG synthesis that involves intrinsic hormonal axes analogous to those operating in the hypothalamic-pituitary-gonadal (HPG) axis. Regulators of pituitary gonadotropins can also influence placental hCG production (at least in vitro). These regulators include progesterone, inhibin, activin, and gonadotropin-releasing hormone (GnRH).
Activin and Inhibin
Activin and inhibin are disulfide-linked homo- and heterodimeric proteins belonging to the transforming growth factor-β (TGF-β) superfamily. Inhibin is a heterodimer composed of an α-subunit and one of two β-subunits, βA or βB. Inhibins (αβA and αβB) derive their name from their ability to preferentially inhibit pituitary FSH secretion. In contrast, activins, composed of βA or βB homodimers (βA-βA and βB-βB), stimulate FSH production. Both hormones affect target cell function via specific cell surface receptors. The bioavailability of activin is restricted by follistatin that binds to activin and prevents it from interacting with its receptor on target cells.
Inhibins are produced by the human placenta. All three subunits are expressed in the syncytiotrophoblast, and the levels of expression do not change with advancing gestation. Activin-A is also produced by the CL, decidua, and fetal membranes during human pregnancy. The placenta also produces follistatin. These factors are secreted into the maternal and fetal circulations and amniotic fluid, and their production varies with stage of gestation.
Although the exact function of the inhibin-activin system in human pregnancy is not known, several studies indicate their involvement in the pathogenesis of gestational diseases. Levels of inhibin-A and activin-A in the maternal circulation can be indicative, albeit with relatively weak predictive value, of pathologies such as placental tumors, hypertensive disorders of pregnancy, intrauterine growth restriction, fetal hypoxia, Down syndrome, fetal demise, preterm delivery, and intrauterine growth restriction.
Because inhibin and activin are synthesized by cytotrophoblast cells, they may be involved in autoregulating placental hCG production by modulating local GnRH activity. In vitro studies have shown that inhibin decreases GnRH-stimulated hCG production by placental cell cultures, whereas inhibin antiserum increases GnRH release and causes a parallel rise in hCG secretion. Thus it is possible that inhibin exerts an autocrine/paracrine effect on placental hCG secretion by suppressing GnRH action. In contrast, activin augments the GnRH-induced release of hCG in cultured trophoblast cells, an effect that can be reduced by the addition of inhibin. Thus, at least in vitro, activin and inhibin, via their paracrine effect on placental GnRH production, contribute to the regulation of hCG secretion in a manner similar to their effect on hypothalamic-pituitary gonadotropin secretion.
Gonadotropin-Releasing Hormone
The human placenta produces GnRH, which is identical to that produced by the hypothalamus. Levels of GnRH in the circulation of pregnant women are highest in the first trimester and correlate closely with hCG levels. The close relation between GnRH and hCG suggests a role for GnRH in regulating hCG production. GnRH stimulates the production of both the α-subunit and β-subunit of hCG in placental explants, and specific GnRH-binding sites are present in the human placenta. Thus there appears to be autoregulation of hCG production within the placenta, suggesting that placental GnRH is essential for the establishment and maintenance of early pregnancy.
Corticotrophin-Releasing Hormone and Urocortin
First identified in the hypothalamus, the corticotrophin-releasing hormone (CRH) is a 41-amino acid peptide that stimulates the expression and processing of proopiomelanocortin (POMC) by pituitary corticotropes and the secretion of adrenocorticotropic hormone (ACTH). The human placenta, fetal membranes, and decidua also express CRH that is identical to that produced by the hypothalamus. Expression of placental CRH can be detected from the seventh week of pregnancy and increases progressively until term, rising more than 20-fold in the last 5 to 7 weeks of pregnancy. Actions of CRH are mediated by two CRH receptors (CRH-Rs): CRH-R1 and CRH-R2. These receptors and various subtypes within each group exhibit tissue-specific expression and possibly contribute to differential actions of CRH on different cell types.
Placental CRH is released mainly into the maternal compartment. Levels of CRH in the maternal circulation can be detected as early as 15 weeks of gestation and then increase through gestation reaching maximum levels of 1 to 10 ng/mL at term. Remarkably, this is about 1000-fold higher than peripheral CRH levels in nonpregnant women.
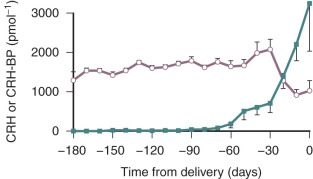
A binding protein (BP) for CRH also exists, and for most of pregnancy, it is present in excess of CRH in the maternal circulation. As the CRH-BP binds CRH with greater affinity than the CRH receptor, it is thought to suppress CRH activity. Thus, for most of pregnancy, the bulk of the placental CRH is considered to be sequestered and rendered inactive by CRH-BP. However, during the last 4 weeks of pregnancy, CRH-BP levels decrease markedly. This coincides with the exponential increase in placental CRH production, which could result in a dramatic increase in CRH biological activity ( Fig. 11.4 ).
Despite the elevated concentrations of CRH during pregnancy, maternal ACTH secretion does not increase concordantly. In fact, pituitary ACTH levels remain low throughout pregnancy. The lack of CRH stimulation could be due to inhibition by the CRH-BP. However, maternal ACTH production remains low late in gestation when CRH increases and CRH-BP decreases. In vivo studies have shown that responsiveness of the maternal pituitary to CRH is markedly attenuated during pregnancy, and in vitro studies have shown that CRH downregulates expression of its receptor in pituitary corticotropes.
In vitro studies indicate that agents that increase CRH production by the hypothalamus also increase CRH production by placental cells. These agents include PGs E 2 and F 2α (PGE 2 and PGF 2α ), norepinephrine, acetylcholine, vasopressin, angiotensin-II, oxytocin (OT), interleukin-I, and neuropeptide-Y (NPY). In contrast, progesterone and nitric oxide donors inhibit placental CRH expression in vitro.
Interestingly, expression and secretion of CRH by the placenta are increased by glucocorticoids. This is in contrast to hypothalamic CRH expression, which is decreased by glucocorticoids. This stimulatory action has been observed in vivo in women who receive glucocorticoid treatment during the third trimester and in vitro in cultured placental cells. The stimulation of placental CRH production by glucocorticoids may result in a positive feedback endocrine loop. Placental CRH may stimulate ACTH production by the fetal pituitary, which would increase cortisol secretion by the fetal adrenals. Fetal adrenal cortisol could then further stimulate placental CRH production. The marked rise in placental CRH during the last 10 weeks of pregnancy could be due to such a positive feedback interaction. This endocrine loop may be involved in the process of parturition (discussed later in the chapter).
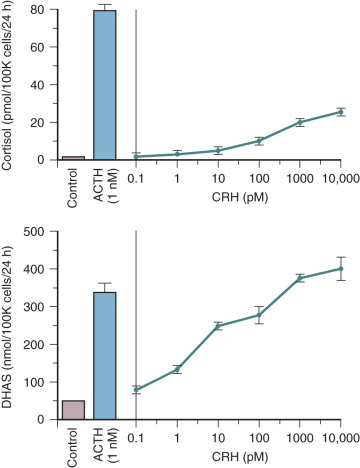
CRH also may influence fetal adrenal steroidogenesis by directly increasing dehydroepiandrosterone sulfate (DHEA-S) production. Interestingly, CRH is as effective as ACTH in stimulating DHEA-S production, but 70% less potent than ACTH in stimulating cortisol production in primary cultures of midgestation human fetal adrenal cortical cells ( Fig. 11.5 ). The capacity for CRH to act as an adrenal androgen secretagogue also has been demonstrated in vivo in adult men. The preferential induction of adrenal androgen synthesis indicates that placental CRH indirectly augments placental estrogen synthesis (discussed later in the chapter). Placental CRH and fetal adrenal DHEA-S increase concordantly during the third trimester, suggesting that production of these two hormones is related. This endocrine axis may play a key role in the regulation of human parturition (discussed later in the chapter).
Several actions have been ascribed to placental CRH in the control of human pregnancy. CRH may serve an autocrine-paracrine function within the placenta by regulating expression and processing of POMC. Placental CRH may be part of the fetal-placental stress response mechanism. The placenta is comparable to the hypothalamus in its production of CRH in response to stress. Neurotransmitters and neuropeptides activated in response to stress stimulate placental CRH release in vitro. The physiologic implications of this are that the fetus may mount a stress response via placental CRH. This may be critical in conditions such as preeclampsia, placental vascular insufficiency, and intrauterine infection.
The CRH-family of neuropeptides also includes a group of structurally and functionally related proteins known as urocortins that have a high affinity for both CRH receptors. The urocortins are expressed by syncytiotrophoblast and extravillous trophoblasts, and by the decidua and fetal membranes. Circulating levels of urocortins are low compared with CRH during pregnancy, suggesting that the peptide acts mainly as a paracrine regulator within the gestational tissue. Urocortin stimulates ACTH and PG production by trophoblast cells and causes vasodilation of the uteroplacental vasculature via activation CRH-R2. In women with decreased uterine artery blood flow during midgestation, circulating urocortin levels are reduced in proportion to the increase in uterine artery resistance. Interestingly, placentas from women with preeclampsia have reduced responsiveness to urocortin, suggesting that a principal role of urocortin is to facilitate vasodilation in the fetoplacental circulation. Studies in animal models also show the vasodilatory effects of urocortin on placental blood flow and suggest that one of its main functions is to protect the fetus from hypoxic insults. Studies of term myometrial cells show that urocortin acting via CRH-R2 increases contractility, suggesting that it plays a role in parturition.
Proopiomelanocortin and Proopiomelanocortin-Derived Hormones
Human placenta expresses POMC. In pituitary corticotropes, this 31-kDa glycoprotein is the precursor for the ACTH-endorphin family of peptides. POMC is enzymatically cleaved into several peptide hormones, including ACTH, β-lipotrophic hormone (β-LPH), α-melanocyte-stimulating hormone (α-MSH), and β-endorphin (β-EP). These neuroendocrine hormones play major roles in the physiologic response to stress and the control of behavior. Each of these peptides, including full-length POMC, has been detected in the human placenta.
The syncytiotrophoblast expresses POMC in a transcriptional pattern, similar to that of extrapituitary tumors. However, the processing of POMC in the placenta is different than that in the pituitary. Although some placental POMC is cleaved, a significant amount of intact POMC is secreted by the placenta into the maternal circulation. In contrast, POMC is processed completely in the pituitary and in nonpregnant adults is undetectable in the circulation. However, during pregnancy, maternal circulating POMC levels are readily detectable by the third month and then increase steadily until midgestation, reaching a plateau of around 300 U/mL between 28 weeks and term. Soon after birth, POMC returns to undetectable levels. Unlike its pattern of secretion by the anterior pituitary, POMC produced by the placenta has no diurnal variability, and it is not inhibited by glucocorticoids. Interestingly, during the third trimester, maternal POMC levels closely correlate with plasma CRH levels but do not correlate with plasma ACTH or cortisol levels.
The physiologic role, if any, of placental ACTH and other POMC-derived proteins in the control of human pregnancy remains to be elucidated. With regard to fetal adrenal growth, placental ACTH plays a negligible role, because it is not sufficient to prevent adrenal hypoplasia in fetal hypopituitarism, due to anencephaly. However, placental ACTH may influence maternal physiology and could be responsible for the relative resistance to negative feedback suppression of pituitary ACTH by glucocorticoids during pregnancy.
Other POMC products are produced by the human placenta. Immunoreactive β-EP in the maternal circulation remains relatively low throughout pregnancy and rises during labor and delivery, indicative of the stress of parturition. Factors that increase pituitary ACTH (e.g., hypoxia and acidosis) also increase β-EP production. Endogenous opioids, enkephalins, and dynorphins are also produced by the placenta. Immunoreactive methionine-enkephalin has been found in the human placenta and is chemically identical to the native molecule. Circulating levels of methionine-enkephalin do not change appreciably throughout pregnancy. Three forms of dynorphin have been found in the human placenta. The amount of dynorphin in the placenta at term is similar to that found in the pituitary gland and brain. Relatively high concentrations of dynorphin are found in amniotic fluid and umbilical venous plasma, and maternal plasma levels in the third trimester, and at delivery are higher than in nonpregnant women. Dynorphin binds to kappa opiate receptors, which are abundant in the human placenta. Dynorphin receptor agonists stimulate the release of human placental lactogen (hPL), suggesting that dynorphin exerts local regulatory effects on hPL production.
Thyrotropin-Releasing Hormone
A substance similar to the hypothalamic thyrotropin-releasing hormone (TRH) has been found in the human placenta. It stimulates pituitary thyrotropin (TSH) release in the rat both in vitro and in vivo, but is not identical to hypothalamic TRH. To date, a placental TSH has not been identified. Whether placental TRH plays a role in stimulating fetal or maternal pituitary TSH remains to be ascertained. The thyroid-stimulating activity of the placenta has been ascribed to hCG.
Placental Lactogen and Somatotropin
In most eutherian mammals the placenta expresses members of the growth hormone (GH)-PL gene family. These genes are encoded by a 66-kb segment of chromosome 17q22-q24, which includes five closely related genes, GH1, GH2, CSH1, CSH2, and CSHL1 , each derived from the duplication of a common ancestral gene. The GH1 gene encodes pituitary GH and is expressed only in the pituitary. The other four are expressed exclusively in the placenta. The GH2 gene encodes a placental GH variant, which differs from pituitary GH by 13 amino acids. The CSH1 and CSH2 genes are identical and encode hPL.
Human Placenta Lactogen
hPL is a single-chain polypeptide of 191 amino acids, with 96% homology with hGH. hPL can be detected in the placenta from around day 18 of pregnancy and in the maternal circulation by the third week of pregnancy. Low levels of hPL (7 to 10 ng/mL) are present in the maternal circulation by 20 to 40 days of gestation. Thereafter, hPL levels in the maternal circulation increase exponentially, reaching levels of 5 to 10 µg/mL at term. In contrast to the elevated levels of hPL in the maternal circulation, concentrations of hPL in the fetal circulation range from 4 to 500 ng/mL at midgestation and only 20 to 30 ng/mL at term. Thus hPL is preferentially secreted into the maternal compartment.
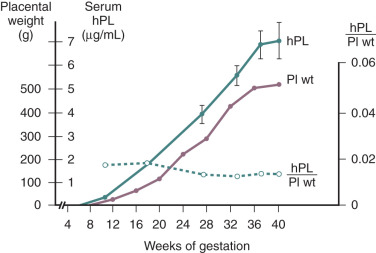
In normal pregnancy, hPL is first synthesized by the cytotrophoblasts of the developing placenta during the first 6 weeks of pregnancy. Thereafter expression switches to the syncytiotrophoblast, which eventually becomes the exclusive source of hPL. The extent of hPL expression by the syncytiotrophoblast does not change during the course of pregnancy, although total placental production increases substantially. Therefore the rise in hPL production is thought to be due to the increase in placental mass, as maternal hPL levels rise concordantly with the amount of syncytiotrophoblast tissue as gestation progresses ( Fig. 11.6 ). After removal of the placenta, the half-life of the disappearance of circulating hPL is 9 to 15 minutes. To maintain circulating concentrations, this would imply placental production of between 1 and 4 g of the hormone per day at term. Thus production of hPL represents one of the major metabolic and biosynthetic activities of the syncytiotrophoblast. hPL is expressed by all types of trophoblastic tissue. It has even been detected in the urine of patients harboring trophoblastic tumors, in men with choriocarcinoma of the testis, and in the serum and urine of women with molar pregnancies.
Factors that regulate hPL production have been assessed in cultured cytotrophoblast cells. Insulin and growth hormone-releasing factor (GRF) stimulate hPL secretion, whereas somatostatin (SS) inhibits its secretion. The presence of hPL, human placental growth hormone (hPGH), SS, and GRF in the same cell suggests that another autoregulatory loop analogous to the hypothalamic-pituitary axis may operate within the placenta. In the third trimester, maternal hPL and GRF levels are closely correlated. Interestingly, SS expression is maximal in early pregnancy and decreases during the second and third trimesters, a pattern opposite to that of hPL secretory activity. Thus locally produced GRF and SS may regulate placental hPL expression. Several studies have demonstrated changes in maternal hPL levels in response to metabolic stress. Specifically, prolonged fasting at midgestation and insulin-induced hypoglycemia raise maternal hPL concentrations. However, hPL levels do not change in association with normal metabolic fluctuations during a typical 24-hour period.
Human Placental Growth Hormone
Two forms of hPGH have been identified, both of which are expressed in the syncytiotrophoblast. The smaller, 22-kDa form is almost identical to pituitary GH, differing by only 13 amino acids. The larger 26-kDa hPGH is a splice variant that retains intron 4. The extent of hPGH production is significantly less than that of hPL, and hPGH is not secreted into the fetal compartment. Consequently, circulating levels of hPGH are approximately 1000-fold less than hPL and can be detected in the maternal circulation later in gestation (between 21 and 26 weeks). During the third trimester, maternal hPGH levels increase exponentially, in concert with hPL, and reach a maximum of approximately 20 ng/mL by term. In the first trimester, pituitary GH is measurable and secreted in a highly pulsatile manner. However, pituitary GH production decreases progressively from about week 15, and by 30 weeks cannot be detected. During the same period, nonpulsatile secretion of hPGH by the placenta increases markedly and becomes the GH of pregnancy.
Biological Actions of hPL and hPGH
Studies of hPL and hPGH deficiency have revealed their potential roles in human pregnancy. In all cases of complete hPL deficiency (lack of detectable hPL in the mother’s blood or in the placenta), pregnancy and fetal development were normal. Thus, despite the remarkably high levels of hPL, it is not essential for normal pregnancy. However, deficiency of both hPL and hPGH due to a mutation in the GH/PL gene cluster results in severe fetal growth retardation, but an otherwise normal pregnancy. Pregnancies in which only hPGH is deficient have not been identified. These experiments of nature indicate that hPL is not necessary for normal pregnancy, whereas normal fetal growth is dependent on either hPGH or hPL, and that these hormones comprise a redundant system.
The initial identification of hPL was based in its lactogenic activity in bioassays, suggesting that hPL acts as a lactogen in human pregnancy. Indeed, hPL binds to the prolactin receptor with relatively high affinity (Kd 0.1 nM). In contrast, its affinity for the GH receptor is low (Kd 770 nM). Thus hPL may function mainly as a lactogen during pregnancy, with only minimal activity as a somatogen (promotes growth). However, administration of hPL to nonpregnant women in sufficient quantities to mimic pregnancy levels did not induce lactation. This does not rule out hPL as a lactogen, as its actions on the mammary gland may be dependent on other factors in the endocrine milieu of pregnancy (e.g., estrogen and progesterone). Nonetheless, the in vivo lactogenic properties, if any, of hPL in human pregnancy remains to be established. It should be noted that maternal prolactin levels increase significantly in the later stages of pregnancy and, together with estrogen and progesterone, are likely sufficient to induce mammary growth and lactation.
As hPGH does not enter the fetal compartment, its principal action is likely on the mother. In contrast, hPL is present in the mother and the fetus. However, deficiency in hPL alone has no effect on fetal growth, whereas deficiency in both hPL and hPGH is associated with severe fetal growth retardation. Taken together, these observations indicate that hPL and hPGH influence fetal growth through effects on the mother. This is consistent with the thesis that hPL and hPGH modulate maternal metabolism to meet fetal energy requirements.
Maternal food intake and intestinal calcium absorption increase during the first trimester, and insulin secretion immediately after feeding almost doubles. Because maternal sensitivity to insulin is maintained in the first half of pregnancy, the increased insulin increases lipogenesis and maternal fat deposition. However, as pregnancy progresses, the fetus increases its nutritional requirements, which leads to an increased functional role for hPL and hPGH. In the second half of pregnancy, maternal cells become increasingly resistant to insulin (i.e., a diabetogenic state), which is thought to be promoted by the combined GH-like and contra-insulin activity of hPGH and hPL. This decreases glucose uptake and increases free fatty acid release. The net effect is increased availability of free fatty acids, glucose, and amino acids for fetal consumption. The decreased maternal glucose consumption would ensure a steady supply of glucose for the fetus, which is especially important for the fetal brain, which uses glucose exclusively as an energy source. Free fatty acids can cross the placenta, and the increased ketones induced by their metabolism are an important energy source for the fetus. Thus, during the second half of pregnancy hPGH and hPL, direct maternal metabolism toward mobilization of maternal energy resources to furnish the needs of the developing fetus. Soon after birth, insulin resistance reverts to the normal nonpregnant state, suggesting that maternal glucose homeostasis is influenced by hormonal factors produced by the fetus-placenta.
This fetal-maternal hormonal interaction represents an example of fetal-maternal conflict whereby the fetus, through natural selection, acquires traits (e.g., placental somatotropin production) that favor the extraction of resources from the maternal organism. Conversely, mothers have evolved mechanisms to counteract fetal demand. Disorders on either side of this equation lead to pathophysiologies of pregnancy. For example, in women with gestational diabetes, insulin secretion is insufficient to balance the decrease in insulin sensitivity, and consequently blood glucose levels increase, leading to an oversupply of glucose to the fetus resulting in macrosomia.
Placental Growth Factors
Expression of many growth factors, cytokines, and their cognate receptors has been detected in the human placenta. Placental growth factors likely play a key role early in pregnancy by affecting implantation, angiogenesis, and vascularization of the implantation site, and the establishment and growth of the placenta. Although many growth factors have been detected in the placenta, their roles in the physiology of pregnancy remain unclear. Some important placental growth factors are discussed below.
Insulin-like Growth Factors
The human placenta produces insulin-like growth factor-I (IGF-I), expressed by the syncytiotrophoblast, and IGF-II, expressed by cytotrophoblasts, syncytiotrophoblast, and extravillous trophoblasts, from as early as week 8 of human pregnancy.
Gene deletion studies in mice have shown that IGF-I and IGF-II are key regulators of placental and fetal growth. The studies suggest that IGF-I is important for fetal and postnatal growth, whereas IGF-II is essential for placental and fetal growth. Pups of IGF-II knockout mice exhibit fetal growth restriction but normal growth after birth, whereas pups of IGF-I knockout mice have poor postnatal growth and die before reaching adulthood. Specific inhibition of placental IGF-II expression inhibits placental and fetal growth, demonstrating the key role of IGF-II during fetal development.
IGFs in the circulation are bound to six GF binding proteins (IGF-BPs) that control IGF function by sequestering circulating IGFs and controlling their bioavailability to interact with receptors on target cells. During pregnancy, IGFBP-1 is produced by the decidua, and all of the IGFBP-1 in amniotic fluid is maternally derived. Overexpression of decidual IGFBP-1 inhibits placental and fetal growth, primarily by sequestering placental IGF-II. Interestingly, IGF-II is an imprinted gene that is expressed only by the paternal allele. In general, genes expressed only by the paternal allele promote fetal/placental growth, whereas those expressed only by the maternal allele suppress fetal growth. Loss of IGF-II imprinting is associated with fetal growth restriction that is thought to be secondary to placental dysfunction. In Beckwith-Wiedemann syndrome, abnormal methylation of the imprinting centers on chromosome 11 leads to additional expression of IGF-II from maternal allele, which is normally suppressed. The extra IGF-II produced in this condition causes fetal macrosomia.
The finding that a paternal gene promotes placental growth supports the concept of genetic conflict between the maternal and fetal genomes. The size and ultimate health of the fetus depends greatly on the size of the placenta. Growth factors that increase placenta size are an advantage to the fetus because they allow it to more efficiently extract resources from the mother. Passage of paternal genes to the next generation is favored if nutrient supply to the fetus is maximized. Maternal genes, on the other hand, not only must survive to the next generation, but they also must ensure that the current pregnancy does not compromise the mother’s future reproductive capacity. Maternal genes (e.g., IGF-BP1) would therefore be selected to oppose the effects of paternally imprinted genes such as IGF-II.
Epidermal Growth Factor Family
The EGF family hormones include EGF, heparin-binding EGF-like growth factor (HB-EGF), amphiregulin, betacellulin, epiregulin, and TGFα. HB-EGF is expressed by the syncytiotrophoblast and cytotrophoblast cells, and this is relatively high in first-trimester and decreases with advancing gestation. EGF-family growth factors are thought to promote trophoblast invasion during implantation and deficiency in EGF expression, and/or signaling is associated with preeclampsia and fetal growth restriction.
Vascular Endothelial Growth Factor Family
The VEGF family of peptides comprises VEGF-A (referred to as VEGF), VEGF-B, VEGF-C, VEGF-D, VEGF-E (generated by alternative splicing from a single gene), and four splice variants of a factor produced by trophoblast cells known as placenta growth factor (PlGF). These peptides are mitogenic and antiapoptotic for endothelial cells and promote the formation of new blood vessels via angiogenesis and vasculogenesis, and increase tissue perfusion via vasodilatation and increased microvascular permeability. Actions of VEGFs and PlGFs are mediated by two tyrosine kinase receptors FMS-like tyrosine kinase-1 (FLT-1) and kinase insert domain receptor (KDR). A soluble form of FLT-1 (sFLT-1) also exists, which is present in the circulation and binds VEGFs and PlGFs to neutralize their angiogenic activity.
Remodeling of the uterine spiral arterioles, and continued angiogenesis through pregnancy to match placental growth, with appropriate maternal vascular supply, are critical for the success of pregnancy. These events are affected by VEGFs and PlGFs expressed by cytotrophoblasts, the syncytiotrophoblast and villous stromal cells in the placenta, and VEGF/PlGF receptor expressed by endothelial cells at the placental/maternal interface. The placental VEGF/PlGF system is linked to pregnancy complications, especially preeclampsia, which is caused by placental factors affecting maternal hemodynamic homeostasis. The etiology of preeclampsia is thought to involve placental hypoxia, due to decreased invasion of trophoblasts into the maternal spiral arteries early in pregnancy, leading to compromised placental perfusion later in gestation, which is thought to induce compensatory mechanisms involving the VEGF/PlGF system to increase placental perfusion by modulating maternal hemodynamics. One mechanism for this is that hypoxia increases placental expression of VEGF, which, via KDR, increases trophoblast expression of sFLT1, leading to increased levels of sFLT1 in the maternal circulation. In pregnancies complicated by preeclampsia, circulating sFLT1 levels in the mother are increased compared with normal pregnancies and sFLT1 levels have biomarker potential to assess risk for preeclampsia and discriminate between normal pregnancies and those that will eventually develop preeclampsia. Excess sFLT1 in the maternal circulation may disrupt maternal endothelium function, leading to the symptoms of preeclampsia, especially hypertension and proteinuria. Thus the placental VEGF/PlGF system appears to play a key role in maintaining maternal vascular function to provide placental perfusion necessary for normal fetal/placental development and the compensatory mechanisms in response to inadequate placental perfusion that can affect the maternal vasculature, leading to complications such as preeclampsia.
Fibroblast Growth Factor Family
Like VEGF, the FGF family (there are 23 members designated FGF-1 to -23) of polypeptides are mitogens that affect endothelial cell migration and promote formation of blood vessels. Effects of FGFs are mediated by four specific high-affinity cell surface FGF receptors (FGFR-1 to -4). The human placenta expresses mainly FGF-2 and FGF-10 that localizes to villous trophoblasts and connective tissue stroma of mesenchymal villi. Interestingly, each of the FGFRs are expressed by Hofbauer cells, mononuclear phagocytes that play a key role in tissue injury repair, inflammation, atherogenesis, and tumor development. FGF induces Hofbauer cells to produce multiple growth factors and cytokines involved in tissue repair. Activation of Hofbauer cells by FGF may promote placental growth by facilitating the outgrowth of syncytiotrophoblast buds. It is also possible that FGF via the placental Hofbauer cells contributes to tissue repair within the placenta during pregnancy.
Placental Adipokines
Adipokines are factors produced by adipose tissue that affect metabolic homeostasis, satiety, and reproduction. Currently known adipokines include leptin, adiponectin, resistin, and ghrelin. Adipokines produced by the placenta regulate the maternal metabolic adaptation to pregnancy, especially increased insulin resistance.
Leptin, a 146-amino acid protein produced primarily by adipocytes, is a key regulator of satiety and body mass index, and its levels are thought to reflect the amount of energy stores and nutritional state. Leptin decreases food intake and body weight via its hypothalamic receptor. In the reproductive system, leptin is thought to coordinate body mass status with reproductive function. In general, leptin acts as a permissive factor. Pulsatile hypothalamic GnRH secretion does not occur unless leptin levels reach a threshold value. Such a mechanism may ensure that energy stores are sufficient to support a pregnancy.
The placenta is the principal source of leptin during pregnancy. Leptin produced by the placenta is secreted mainly into the maternal circulation, and as a consequence leptin levels are elevated during pregnancy. In the first trimester, maternal plasma leptin levels double nonpregnant values and continue to increase during the second and third trimesters, during which time they are also expressed by the chorion and amnion. Leptin levels decline to normal nonpregnant values within 24 hours of delivery.
The influence of placental leptin on maternal biology is unclear. Abnormally high placental leptin production is associated with maternal diabetes mellitus and hypertension, and umbilical leptin levels correlate with fetal adiposity. Interestingly, leptin levels during pregnancy do not correlate with body mass index as they do in the nonpregnant state. Pregnancy appears to be a state of hyperleptinemia and leptin resistance, with uncoupling of leptin effects on eating behavior, satiety, and metabolic activity. Leptin is lipolytic and favors fatty acid mobilization from adipose tissue. It also may act in the liver, pancreas, and muscle to decrease insulin sensitivity and mobilize glucose. The human placenta expresses leptin receptors, and therefore leptin can act in a paracrine manner to modulate placental function. Leptin induces hCG production in trophoblast cells and is also thought to increase placental growth by augmenting mitogenesis, amino-acid uptake, and extracellular matrix synthesis.
Adiponectin, resistin, and ghrelin are also produced by the placenta and secreted into the maternal and fetal compartments. As with leptin, these adipokines appear to affect maternal metabolic homeostasis in favor of pregnancy and the supply of nutrients to the fetus.
Placental Extracellular Vesicles
Villous trophoblasts and the syncytiotrophoblast produce extracellular vesicles (EVs; aka exosomes, microparticles) that contain biologically active signaling factors in the form of nucleic acids (messenger RNA, microRNA [miRNA], and noncoding RNA), proteins and lipid. Exosomes, 30 to 100 nm vesicles, are produced by trophoblast cells and shed via endosomal trafficking primarily into the maternal compartment. Placental exosomes exert biological effects by fusing with the target cell plasma membrane and depositing their cargo of bioactive factors into the cytoplasm. Placental exosomes may regulate proximal and distal targets and represent key vectors for communication from the fetus/placenta to the mother. They can been detected in the maternal circulation from the sixth week of gestation, and their levels increase gradually with advancing gestation in proportion to the increased size of the placenta. miRNAs in placental exosomes affect the function of local immune cells to boost resistance to viral infection. In addition, placental exosomes carry factors to the maternal compartment that affect maternal immune tolerance toward the fetus at the maternal-fetal interface. Thus placental EVs, mainly in the form of exosomes, mediate communication between that fetus and mother via the packaging of a specific cargo of signaling molecules that are protected from degradation in the circulation, to exert specific effects on target cell in the mother.
Levels of exosomes (placental and nonplacental) in the maternal circulation and the composition of their cargo has been associated with pregnancy complications, such as preeclampsia, preterm birth, intrauterine growth restriction, and gestational diabetes. This suggests that the content of placental exosomes are not only involved in the physiology of pregnancy, but also could be used as biomarkers to predict specific pregnancy complications.
Trophoblast EVs also contain fragmented (~200 base pairs in length) fetal DNA into vesicular that are shed into the maternal circulation. The DNA is referred to as cell-free fetal DNA (cffDNA). cffDNA originates in trophoblast cells and can be assayed in the maternal blood. cffDNA is detectable in the maternal circulation from around the seventh week of gestation, and the amount increases with advancing gestation, such that late in pregnancy it represents ~4% of the cell-free DNA in maternal blood. After delivery of the placenta, the amount of cffDNA in maternal blood rapidly decreases. The physiologic role of cffDNA and why it is packaged into vesicles that are shed from trophoblast cells into the maternal compartment is not known. Because it can be isolated and sequenced, cffDNA is used for noninvasive prenatal diagnosis and testing for conditions such as X-linked genetic disorders and aneuploidies, as well as fetal sex determination.
Placental Steroid Hormones and the Fetal-Placental Unit
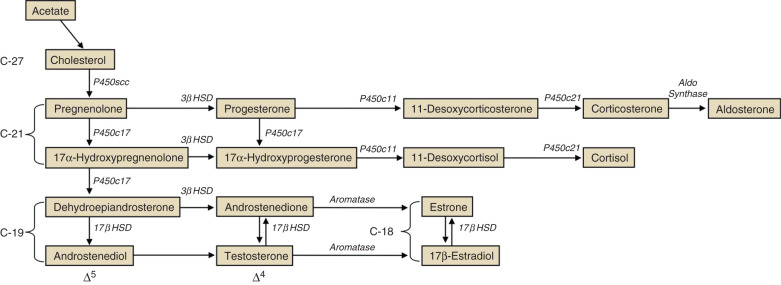
Steroid hormone production and metabolism by the placenta is dependent on precursors provided by the fetus and mother. The interdependence of fetus, placenta, and mother for steroid hormone production in human pregnancy led to the concept of an integrated fetal-placental-maternal steroidogenic unit. To understand this concept, it will be useful to review the general steroid biosynthetic pathways ( Fig. 11.7 ; also discussed in Chapter 4 ).
Progesterone
The human placenta produces large amounts of progesterone throughout pregnancy. It does this mainly by converting low-density lipoprotein cholesterol, extracted from the maternal circulation, to pregnenolone, which is then converted to progesterone. Cholesterol is converted to pregnenolone in the mitochondria of trophoblast cells by the cytochrome P450scc enzyme. This is the rate-determining step for placental progesterone production. The conversion of pregnenolone to progesterone also occurs in the mitochondria and is catalyzed by type-1 3β-hydroxysteroid dehydrogenase (3βHSD-I). The human placenta is incapable of converting progesterone to 17α-hydroxyprogesterone. Production of progesterone approximates 250 mg/day by the end of pregnancy, at which time circulating levels are on the order of 130 ng/mL.
As its name implies, progesterone is a progestation hormone and has been aptly called the “hormone of pregnancy” because it is essential for the establishment and maintenance of pregnancy. In all viviparous animals examined so far, disruption of progesterone synthesis or action during pregnancy induces pregnancy termination or labor and delivery. The role of progesterone in the maintenance of pregnancy and the mechanism by which its actions are withdrawn to trigger parturition are discussed later in this chapter.
Estrogens
The principal role of estrogens in human pregnancy is to stimulate uterine growth and increase uterine blood flow. Estrogens also affect breast development in preparation for lactation. At parturition, estrogens oppose the actions of progesterone by augmenting uterine contractility and inducing cervical softening.
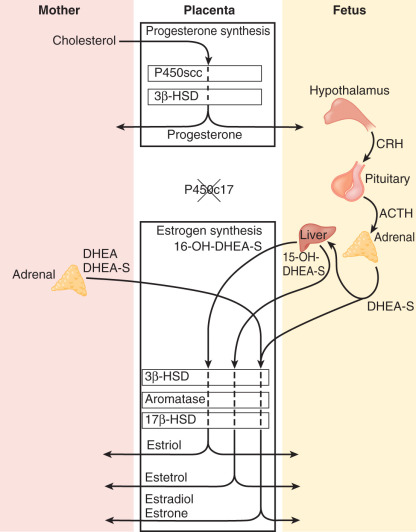
Estrogens are synthesized by the human placenta from C19 steroids. The principal precursor used for placental estrogen formation is DHEA-S, supplied mainly by the fetal adrenal glands. Because the placenta has an abundance of the sulfatase (sulfate-cleaving) enzyme, DHEA-S is rapidly converted to free (unconjugated) DHEA, which is then converted to androstenedione by 3βHSD-I. The human placenta also expresses high levels of the aromatase enzyme, which converts androstenedione estrone. The 17β hydroxysteroid dehydrogenase (17βHSD) enzymes then interconvert estrone and estradiol ( Fig. 11.8 ).
The major estrogen formed during human pregnancy is estriol, which has an additional hydroxyl group at position 16. Estriol constitutes more than 90% of the estrogen in pregnancy urine, into which it is excreted as sulfate and glucuronide conjugates. Estriol production by the placenta increases with advancing gestation, and ranges from approximately 2 mg/24 hours at 26 weeks to 35 to 45 mg/24 hours at term. At term, the concentration of estriol in the maternal circulation is 8 to 13 ng/dL. In contrast, ovarian production of estriol in nonpregnant women is barely detectable.
Placental estriol is formed by a biosynthetic process unique to human (and higher primate) pregnancy, that begins with DHEA-S from the fetal adrenal glands. When DHEA-S of either fetal or maternal origin reaches the placenta, estrone and estradiol are formed. However, little of either is converted to estriol by the placenta. Instead, some of the DHEA-S undergoes 16α-hydroxylation to form 16α-hydroxyDHEA-S, primarily in the fetal liver and, to a limited extent, in the fetal adrenal. In the placenta, that sulfatase enzyme converts 16α-hydroxyDHEA-S to 16α-hydroxyDHEA, which is then aromatized to form estriol. Thus levels of estriol in the maternal blood reflect the steroidogenic activity of the fetal HPA. In the mother, estriol is conjugated to form estriol sulfate and estriol glucosiduronate in the liver and excreted via the urine. The function of estriol in pregnancy has attracted much speculation. In most biological systems, estriol is a weak estrogen, with approximately 1% the potency of estradiol and 10% that of estrone. However, in pregnancy, the uterus estriol is as effective as the other estrogens in increasing uteroplacental blood flow.
Another placental estrogen derived from a fetal precursor is estetrol, formed after 15 and 16-hydroxylation of DHEA-S. Its function is not known. Relative levels of progesterone and estrogens (estrone, estradiol, estriol, and estetrol) in the maternal circulation during human pregnancy are shown in Fig. 11.9 .
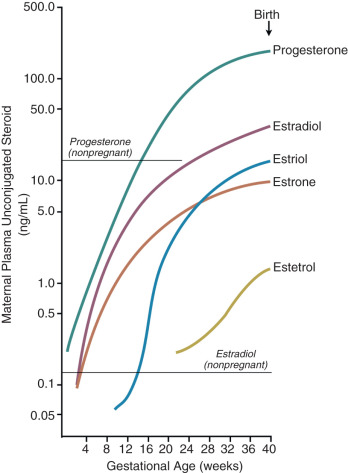
Production of estrogens by the human placenta is extraordinarily high, and circulating levels during pregnancy are several orders of magnitude higher than physiologic levels in nonpregnant women. A key question, therefore, is why does the human placenta make so much estrogen? Conditions in which placental estrogen synthesis is markedly decreased (e.g., anencephaly, congenital adrenal lipoid hyperplasia, placental aromatase deficiency, and placenta sulfatase deficiency) have provided some insight into the role of the placental estrogens in human pregnancy. Although pregnancy was prolonged in some cases of placental sulfatase deficiency and anencephaly, in most cases, a marked decrease in placental estrogen synthesis had little effect on fetal and placental development and the timing of parturition. Likewise, pregnancies with lowered estrogen levels due to placental aromatase deficiency appear normal, although mothers and female fetuses exhibited virilization signs due to androgen excess. Thus high levels of estrogens of placental origin may not be essential for normal pregnancy and parturition, but rather a critical role of placental aromatase is to metabolize circulating androgens to protect the female fetus and mother from virilization.
These observations do not exclude a role for estrogen in the control of human pregnancy. Although levels of maternal estrogens in pregnancies with placental estrogen synthesis defects were low compared with normal pregnancies, they were still in a physiologically significant range (1 to 1.6 nmol/L) and comparable with levels reached in the midcycle and luteal phase of the menstrual cycle (0.6 to 2 nmol/L). Thus, despite the inability of the placenta to produce estrogens in certain abnormalities such as aromatase deficiency, the estrogen target tissues were still exposed to moderately high levels of estradiol. This suggests that a minimal level of estrogen is necessary for human pregnancy, and that the excessive levels produced by the placenta are redundant.
Placental Glucocorticoid Metabolism
For most of gestation the placenta expresses the 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD-2) enzyme, which inactivates cortisol by catalyzing its conversion to cortisone. As maternal cortisol levels are 3 times those of the fetus, this biochemical barrier serves to prevent excess cortisol from entering the fetal compartment. This is important because exposure of the fetus to high levels of cortisol not only could interfere with the fetal hypothalamic-pituitary axis, but also is associated with decreased birth weight and hypertension.
Studies in the baboon placenta suggest that estrogen upregulates 11β-HSD-2 expression in the placenta. Because estrogen synthesis by the placenta requires fetal adrenal androgen precursors (produced in part by fetal pituitary ACTH stimulation), the estrogen regulation of placental 11β-HSD-2 represents a regulatory loop that ensures that maternal cortisol does not affect the fetal HPA axis.
Fetal Neuroendocrine Systems
- ◆
Perinatal survival and future health of the newborn is dependent on the ordered functional maturation of fetal neuroendocrine systems in preparation for the neonate to maintain homeostasis and adapt to the extrauterine environment.
The appropriate functional maturation of neuroendocrine systems during fetal life has profound effects on long-term health into adulthood. This phenomenon, referred to as developmental origins of adult health and disease, was first observed in epidemiologic studies, showing an association between perturbations of intrauterine nutritional status and the development of hypertension, insulin resistance, and obesity that predispose to cardiovascular disease, diabetes, and the metabolic syndrome in adulthood.
The developmental origins hypothesis posits functional plasticity in the development of fetal neuroendocrine and organ systems, such that a single genotype gives rise to multiple “normal” phenotypes in response to environmental cues (e.g., extent of maternal nutrition and stress). Thus the fetus prepares itself for the extrauterine environment by modulating its physiologic systems to match the anticipated extrauterine environment based on maternal cues. Interestingly, neuroendocrine plasticity does not continue later in life. The period of neuroendocrine plasticity is limited to fetal and possible early neonatal development, and the physiologic changes appear to be permanent. For example, studies in animals show that changes in basal and stress-associated fetal HPA activity induced in utero by maternal starvation persist after birth. The altered stress-response pathway may underpin pathophysiologic conditions that develop during adulthood.
The physiologic, biochemical, and genetic mechanisms that mediate the reactive plasticity of fetal neuroendocrine development are not clearly defined. The placenta is thought to play an active role, although it remains uncertain how information regarding the environment is transmitted from the mother, through the placenta and to the fetus. As described previously, the placenta produces a plethora of hormones that could influence the developmental trajectories of fetal neuroendocrine axes. For example, placental CRH may modulate the responsiveness of the fetal adrenal cortex to ACTH and therefore contribute to the establishment of the HPA set point.
It is also possible that perturbations in the gestational milieu induce static changes in chromatin structure, referred to as epigenetic modifications that alter gene expression and persist into adult life. The most common epigenetic modifications (also known as marks) are methylation of cytosines in CpG dinucleotides and posttranslational modifications (e.g., acetylation) of histones. Methylation in gene promoter regions generally inhibits downstream gene expression, and histone modifications alter the chromatin secondary structure, affecting activity of the transcriptional machinery. These chemical modifications do not involve changes in the primary DNA sequence and are heritable by mitosis from one cell generation to the next. During the preimplantation period, embryonic cells undergo waves of demethylation that deprogram all epigenetic marks to generate cells with broad developmental potential. The DNA is then remethylated as the embryo develops. This provides an opportunity for environmental influences to affect the epigenome (genome-wide pattern of epigenetic marks). Epigenetic modification during development is thus a plausible mechanism for environment-induced phenotypic plasticity and provides a mechanistic explanation for the effects of prenatal and perinatal environmental exposures (e.g., malnutrition) on the function of neuroendocrine systems and their effect on subsequent disease risk in later life.
Current understanding of the developmental patters of the major neuroendocrine systems in the human fetus is discussed below.
Hypothalamic Hormones
By the end of the fifth week of pregnancy, the primitive hypothalamus can be identified as a swelling on the inner surface of the diencephalic neural canal. It then differentiates to form a complex of interconnecting nuclei. By 9 to 10 weeks, the median eminence of the hypothalamus is evident, and interconnecting fiber tracts of hypothalamic nuclei can be identified by 15 to 18 weeks. The hypophysiotropic hormones GnRH, TRH, CRH, growth hormone-releasing hormone (GHRH), and SS appear in the fetal hypothalamus during this period ( Table 11.2 ).
| Hormone | Age Detected (wk) |
|---|---|
| Hypothalamic | |
| Gonadotropin-releasing hormone | 14 |
| Thyrotropin-releasing hormone | 10 |
| Somatostatin | 14 |
| Dopamine | 11 |
| Growth hormone–releasing hormone | 18 |
| Corticotropin-releasing hormone | 16 |
| Pituitary | |
| Prolactin | 16.5 |
| Growth hormone | 10.5 |
| Corticotropin (ACTH) | 7 |
| Thyroid-stimulating hormone (thyrotropin) | 13 |
| Luteinizing hormone | 10.5 |
| Follicle-stimulating hormone | 10.5 |
Gonadotropin-Releasing Hormone
The human fetal hypothalamus produces immunoreactive and bioactive GnRH by 10 weeks of gestation. Between 10 and 22 weeks, the concentration of GnRH in the fetal hypothalamus remains constant (0.27 to 13.1 pg/mg) and is not different between the sexes. GnRH release from human fetal hypothalamic explants is pulsatile. In male rhesus monkey fetuses, the pituitary-gonadal axis is active during the last third of gestation. The pituitary produces LH in response to GnRH, and the testes produce testosterone in response to LH. A similar situation likely exists in the human male fetus. Whether this also is the case for the female fetus is unresolved.
Thyrotropin-Releasing Hormone
Significant levels of immunoreactive TRH are found in the human fetal hypothalamus early in gestation. As with GnRH, fetal hypothalamic TRH levels do not correlate with sex or gestational age. The presence of TRH in the fetal hypothalamus in early gestation and midgestation suggests its role in the regulation of TSH, and possibly prolactin (PRL), secretion.
Growth Hormone-Releasing Hormone and Somatostatin
GHRH can be detected in fetal hypothalamic neurons and fiber tracts at 18 weeks, with increasing levels found up to 30 weeks. The simultaneous detection of GHRH in neuron cell bodies and fiber tracts indicates its release into portal vessels by midgestation. Immunoreactive SS can be identified in the hypothalamus of human fetuses from 10 to 22 weeks. In contrast to GnRH and TRH, fetal hypothalamic SS increases with advancing gestational age.
Corticotropin-Releasing Hormone and Arginine Vasopressin
CRH is a potent secretagogue for ACTH and β-endorphin release by the human fetal pituitary gland. Arginine vasopressin (AVP) also directly stimulates ACTH secretion by the fetal pituitary and can synergize with CRH. CRH-immunoreactive fibers can be detected in the median eminence between 14 and 16 weeks. CRH immunoactivity and bioactivity and AVP immunoactivity have been detected in human hypothalamic extracts from 12 to 13 weeks.
Hypothalamic CRH and AVP increase with gestational age, and the CRH bioactivity of fetal hypothalamic extracts, measured in isolated rat anterior pituitary cells, is augmented by AVP. Thus the human fetal hypothalamus has the capacity to regulate pituitary ACTH production from early in the second trimester. The extent to which fetal hypothalamic CRH and AVP and placental CRH interact in regulating fetal ACTH release remains uncertain.
Catecholamines and Dopamine
Catecholamines in cells projecting from the arcuate nuclei to the internal and external layers of the median eminence appears during the interval from 12 to 16 weeks. Dopamine is present in the fetal hypothalamus at weeks 11 to 15 at a concentration twice that of the adult. Hypothalamic dopamine inhibits PRL release from fetal the pituitary during this time.
Pituitary Hormones
The pituitary primordium appears as the epithelial evagination of Rathke’s pouch, arising from a diverticulum of the stomadeum. The evaginated pituitary primordium appears at 4 weeks and separates from the stomadeum by 5 weeks. The floor of the sella turcica is in place by week 7 and separates the pituitary from its epithelial origins. The progenitors of pituitary hormone-secreting cells originate in the ventral neural ridges of the primitive neural tube. This region also gives rise to the diencephalon, suggesting that the hypothalamus and anterior pituitary share a common embryonic origin.
The pituitary increases in size and cell number through proliferation of cell cords into mesenchyme, beginning during week 6. Capillaries interdigitate among the mesenchymal tissue of Rathke pouch and the diencephalon at 8 weeks, and the median eminence is distinguishable by 9 weeks. The hypothalamic-hypophyseal vascular system in fetuses is intact by 11 to 16 weeks.
The anterior pituitary gland comprises five types of specialized epithelium-derived secretory cells: (1) lactotropes producing PRL, (2) somatotropes producing GH, (3) corticotropes producing ACTH, (4) thyrotropes producing TSH, and (5) gonadotropes producing LH and FSH.
Within the pituitary, cells containing ACTH have been detected at 7 weeks, β-lipotropin-containing and β-endorphin-containing cells at 8 weeks, GH-containing and LH-containing cells both at 10.5 weeks, and TSH-containing cells at 13 weeks. MSH-containing cells, which probably contain β-lipotropin, have also been reported to appear at 14 weeks, and PRL-containing cells appear at 16.5 weeks (see Table 11.2 ). Many of the components required for the normal regulated function of the secretory cells of the anterior pituitary, the hypophysiotropic factors elaborated by the hypothalamus, and the neurohemal link connecting the hypothalamus and pituitary are present in the fetus well before the end of the first half of pregnancy. The primate fetal pituitary is competent to respond in vitro to virtually all of the known hypophysiotropic factors by midgestation. Whether it responds to these compounds in vivo remains to be demonstrated directly, although studies in the catheterized rhesus monkey fetus in utero suggest that responses to at least one of the hypophysiotropic factors, GnRH, occur.
Fetal Pituitary-Adrenal Axis
The anlage of the human adrenal cortex is first identified at about the fourth week of gestation as a thickening of the celomic epithelium in the notch between the primitive urogenital ridge and the dorsal mesentery. By the fifth week, these primitive cells begin to migrate toward the cranial end of the mesonephros, where they condense to form the earliest recognizable manifestation of the adrenal gland. Cells destined to become the steroidogenic cells of the adrenal and gonad are derived from neighboring areas of the celomic epithelium and are morphologically identical. In general, the portion medial to the mesonephros produces cells destined for the adrenal cortex, whereas the portion ventral to the mesonephros produces cells destined for the gonad.
During the last two-thirds of gestation in humans and higher primates, the fetal adrenal glands are disproportionately enlarged and exhibit extraordinary growth and steroidogenic activity. The growth is attributable to cortical hypertrophy controlled by locally produced growth factors, including basic FGF and IGF-II, in response to ACTH.
For much of gestation, the human fetal adrenal cortex is composed of two morphologically distinct zones, the fetal zone and the definitive zone. The fetal zone accounts for 80% to 90% of the cortex and is the primary site of growth and steroidogenesis. During midgestation, the fetal zone produces 100 to 200 mg/day of DHEA-S, which is quantitatively the principal steroid product of the human fetal adrenal gland throughout gestation. As its name implies, the fetal zone exists only during fetal life; it involutes or undergoes remodeling soon after birth. Clusters of immature neuroblasts are present between the innermost fetal zone cells. These cells aggregate at birth to form a functional adrenal medulla.
The definitive zone (also referred to as the adult cortex, neocortex, or permanent zone) comprises a narrow band of small, tightly packed basophilic cells that exhibit structural characteristics typical of cells in a proliferative state. Its inner layers form arched cords that send fingerlike columns of cells into the outer rim of the fetal zone. Definitive zone cells begin to resemble steroidogenically active cells later in pregnancy, and appear to secrete mineralocorticoids late in the third trimester.
Ultrastructural and functional studies have revealed a third zone between the fetal and definitive zones, referred to as the transitional zone, the cells of which have intermediate characteristics between the fetal and adult zones. After midgestation, transitional zone cells have the capacity to synthesize cortisol and thus may be analogous to cells of the zona fasciculata of the adult adrenal. By week 30 of gestation, the definitive zone and transitional zone begin to take on the appearance of the zona glomerulosa and the zona fasciculata, respectively. Thus, by late gestation, the fetal adrenal cortex resembles a rudimentary form of the adult adrenal cortex.
The postnatal remodeling of the primate adrenal cortex involves a complex wave of differentiation, such that the inner portion of the fetal zone atrophies and the zonae glomerulosa and fasciculata develop. Fetal zone remodeling in the human is an apoptotic process. It has generally been thought that the adult cortical zones develop from the persistent definitive zone. However, there is no evidence of adrenal cortical insufficiency during the neonatal period when the postnatal remodeling process is at its peak. It is more likely that the nascent adult cortical zones are present and functional before birth. Indeed, morphologic and functional studies have identified rudimentary zonae glomerulosa and fasciculata during late gestation.
ACTH secreted from the fetal pituitary is the principal trophic regulator of the fetal adrenal cortex. However, ACTH may not be acting directly. During the last two-thirds of gestation, the fetal zone grows rapidly and produces large amounts of steroids, even though circulating ACTH concentrations do not rise significantly. Soon after birth, the fetal zone rapidly involutes but exposure to ACTH continues, albeit at lower concentrations. Other factors, possibly specific to the intrauterine environment, appear to play a role in the regulation of fetal adrenal cortical growth and function. Substances produced by the placenta (e.g., hCG) have been implicated, and peptide growth factors produced locally within the fetal adrenal appear to influence fetal adrenal cortical growth and function by mediating or modulating the trophic actions of ACTH.
Evidence of fetal adrenal steroidogenesis is first seen at 6 to 8 weeks, when the cells in the developing adrenal differentiate and acquire steroidogenic characteristics. At around week 12 of gestation, estriol concentrations in the maternal circulation rapidly increase (~100-fold). This increase coincides with the initiation of fetal zone enlargement and ACTH secretion by the fetal pituitary gland. Production of DHEA-S by the fetal adrenal cortex continues for the remainder of pregnancy and increases considerably during the second and third trimesters. By term, the human fetal adrenal produces around 200 mg DHEA-S per day. Thus placental production of estriol directly reflects the steroidogenic activity of the fetal HPA axis. For this reason, maternal estriol previously was used as an endocrine marker to evaluate fetal well-being.
The point at which the fetal adrenal cortex begins producing physiologically relevant amounts of cortisol has yet to be determined definitively. This question has been partially answered, however, by observations of infants with congenital adrenal hyperplasia (CAH). In CAH, due to a deficiency of the 21-hydroxylate (P450c21) enzyme, the fetal adrenals cannot synthesize cortisol. As a result, excess ACTH is produced due to loss of glucocorticoid-negative feedback. Female infants with CAH often are born with urogenital sinus defects and virilization, primarily caused by exposure of the urogenital sinus to excessive amounts of adrenal androgen. Because this condition derives from adrenal glucocorticoid deficiency, the observations imply that the adrenal produces enough cortisol in female fetuses before 10 weeks of gestation to regulate ACTH levels and to prevent overproduction of adrenal androgens that may masculinize the female urogenital sinus. Expression of key steroid metabolizing enzymes suggests that the human fetal adrenal cortex does not produce cortisol de novo from cholesterol until around week 30 of gestation. However, this does not preclude the possibility that cortisol is produced by using progesterone as a precursor early in gestation.
Because the fate of pregnenolone metabolism is initially determined by the branch point steroidogenic enzymes P450c17 and 3β-HSD, the steroidogenic potential of cells may be inferred by the pattern of expression of these two enzymes (see Fig. 11.7 ). Expression of 3β-HSD by the human fetal adrenal cortex is a critical step in the metabolism of pregnenolone, because it confers on cells the ability to convert Δ 5 -3β-hydroxysteroids to Δ 4 -3 ketosteroids essential for mineralocorticoid and glucocorticoid production. Between 12 and 22 weeks of gestation, the human fetal adrenal cortex does not express 3β-HSD. After 22 weeks, 3β-HSD expression can be detected first in the definitive zone cells and later in gestation in the definitive and transitional zone cells. At no time in gestation is 3β-HSD expression detected in the fetal zone. In contrast, expression of P450c17 is highly abundant in the transitional and fetal zones and is lacking in the definitive zone at all gestational ages.
The persistent lack of 3β-HSD and expression of large amounts of P450c17 in the fetal zone is consistent with this cortical compartment producing only C19/Δ 5 steroids, particularly DHEA. The lack of P450c17 in the definitive zone, and the eventual expression of 3β-HSD in this compartment, is consistent with this zone producing mineralocorticoids late in gestation. The coexpression of 3β-HSD and P450c17 in the transitional zone indicates that this zone has the capacity for glucocorticoid production ( Fig. 11.10 ).
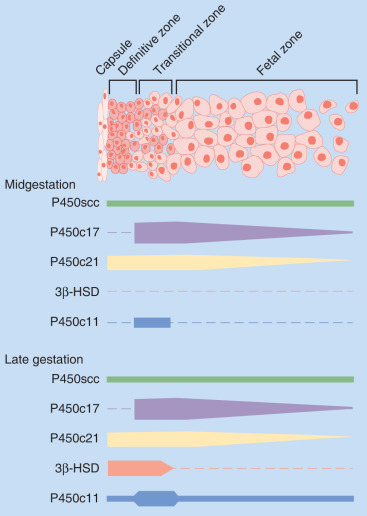
Related posts:
 Gonadotropin Hormones and Their Receptors
Gonadotropin Hormones and Their Receptors
 Steroid Hormones and Other Lipid Molecules Involved in Human Reproduction
Steroid Hormones and Other Lipid Molecules Involved in Human Reproduction
 Structure, Function, and Evaluation of the Female Reproductive Tract
Structure, Function, and Evaluation of the Female Reproductive Tract
 Hormone-Responsive Cancers
Hormone-Responsive Cancers
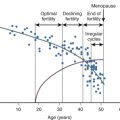 Medical Approaches to Ovarian Stimulation for Infertility
Medical Approaches to Ovarian Stimulation for Infertility
 Pelvic Imaging in Reproductive Endocrinology
Pelvic Imaging in Reproductive Endocrinology
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


