Endocrine neoplasms are unique among neoplasias of other body systems in that the majority of them have a very benign appearance under the microscope. Additionally, many of these lesions are long-standing and, unless they are hormone-producing or obstructive, with either leading to clinical symptoms, they often are found incidentally. Unfortunately, as is the case for many endocrine neoplasms, the true marker of malignancy is metastasis. However, a good deal of progress has been made in the molecular pathophysiology of endocrine tumors.
Cancer of the Thyroid Gland
Thyroid carcinomas, of all carcinomas, have the highest increase in incidence at 5.3% per year (1995–2004 SEER data) and the second highest increase in U.S. cancer deaths (0.6% per year). In 2007 there were 35,520 expected new cases of thyroid carcinoma, and these changes included a female-to-male incidence of 3:1 and 1:1 female-to-male death rates. Those grim statistics said, the 5-year overall survival rate with thyroid cancer among men and women is over 97%. This high rate of survival may be reflective of early symptoms in truly aggressive lesions or increased diagnosis due to less clear molecular features of malignancy. Cancer of the thyroid gland is a relatively uncommon malignancy, marked by slow growth, delayed symptoms, and a low incidence of morbidity and mortality. In the United States approximately 17,000 new cases are diagnosed each year, and about 1700 deaths occur. The incidence of thyroid cancer in women is twice that in men, with a peak occurring in the third and fourth decades. The prevalence of cancer in a solitary nodule is approximately 1%. The prevalence of microscopic foci of unsuspected thyroid malignancy in multinodular glands is approximately 10% to 15%.
The most well documented risk factor for thyroid cancer is radiation administered during childhood for benign conditions of the head and neck or thymus. The risk seems to be dose-related, with the greatest risk between 2 Gy and 10 Gy. Thyroid cancer is thought to be less common after doses higher than 20 Gy, probably because few cells remain to undergo malignant degeneration after radiation-induced atrophy and fibrosis. However, thyroid cancer cases have been reported within the atypical nodular hyperplasia of the thyroid occurring after radiation for childhood Hodgkin disease or rhabdomyosarcoma of the neck. Papillary and follicular cancers are most commonly associated with prior radiation exposure. Other less well defined risk factors for thyroid cancer include endemic goiter, high iodine intake, Graves’ disease, and Hashimoto’s disease (papillary). 131 I therapy for hyperthyroidism does not appear to increase the risk of thyroid cancer. Medullary carcinoma of the thyroid can be sporadic or inherited as an autosomal-dominant trait, either alone or in the context of multiple endocrine neoplasia (MEN) type II. Patients with Hashimoto’s thyroiditis also are at increased risk for lymphoma of the thyroid.
HISTOLOGIC SUBTYPES AND FEATURES
Table 11.1 lists the various subtypes of malignant tumors of the thyroid gland.
| Epithelial Tumors | Nonepithelial and Miscellaneous Tumors |
|---|---|
| Papillary thyroid carcinoma | Lymphoma |
| Follicular carcinoma | Sarcoma |
| Poorly differentiated carcinoma | Metastatic tumor |
| Undifferentiated (anaplastic) carcinoma | Unclassified malignant neoplasm |
| Medullary carcinoma | |
| Squamous cell carcinoma |
Papillary Carcinoma
The most common malignancies of the thyroid gland arise within the glandular epithelium, and as such, are adenocarcinoma (carcinomas associated with glandular epithelium). In pathologic diagnoses the “adeno” is typically not used, because inherent carcinomas of the thyroid gland are adenocarcinomas.
Papillary thyroid carcinomas (PTCs) occur in highest incidence, and these represent more than 70% of adult thyroid cancers. PTCs most often arise in young adults and show a predilection for females (75%).
Diagnostic pathologic features of PTC include cellular crowding, cleared-out nuclei with dispersed chromatin and occasionally prominent nucleoli (“Orphan Annie eyes”), irregular nuclear borders, loosely assembled, resulting in characteristic nuclear grooves, and intranuclear pseudoinclusions. There are numerous subtypes (variants) of PTC, including classical, follicular, tall cell, diffuse-sclerosing, and solid types, just to name a few of the more commonly encountered variants. These PTC subtypes vary widely in terms of obvious nuclear features and prognoses.
More recently it has become widely recognized through improved imaging modalities that the incidence of thyroid nodules is much greater than the 10% at autopsy often quoted in the literature and texts. Biopsy of these subcentimeter nodules has led to a rapid increase in the diagnosis of PTC. Furthermore, it is unclear that these microscopic, clinically silent nodules would ever progress to lethality. They are pathologically referred to as papillary thyroid microcarcinomas and thought to be of low biologic significance. However, these microscopic foci have been shown to harbor the same genetic alterations as their more lethal relatives, making their true significance unclear. There is some subjectivity, and clinical experience seems to play a role in prognostication. For instance, a microscopic focus of what would be considered to be a higher-grade subtype of PTC, such as tall cell or diffuse sclerosing variant, would probably be regarded to be of greater biologic significance on the basis of the more aggressive clinical course of these carcinomas when they are discovered at larger than 1 cm, the size of tumor considered to be a true pathologic carcinoma by the World Health Organization (WHO) classification system.
Traditionally, involvement of local lymph nodes has been a common finding in PTC, although increased earlier detection has lowered this statistic. The presence of local lymph node metastases, however, does not portend a worse prognosis. Less than 10% of PTCs have metastasized outside the neck region at the time of presentation, the most common sites of metastases being lung and bone. The overall 10-year survival rate is about 93%. Primary treatment is surgery. Adjuvant treatment is often radioactive iodine, imported by thyrocytes via the sodium-iodide symporter, causing local destruction of any thyroglobulin-producing cells (as well as neighboring cells), a method of combating non–hormone-producing cells with clinically local disease. Pretreatment adjuvant therapy with human recombinant thyroid-stimulating hormone (TSH) has been used in some centers to improve uptake of radioactive iodine.
Follicular Carcinoma
Follicular-patterned lesions are a diagnostic conundrum. In that fine-needle aspiration via ultrasound guidance has become the diagnostic modality of choice for primary solitary or multiple large nodules of the thyroid, follicular lesions can present a unique challenge. Among the follicular-patterned lesions are follicular adenoma (FA), follicular carcinoma (FC), PTC (follicular variant), follicular nodules of multinodular goiter, Graves’ disease, and Hashimoto’s thyroiditis. Although the autoimmune thyroiditides and multinodular goiter can present with follicular-patterned nodules, the real diagnostic dilemma is distinguishing among FA, FC, and sometimes PTC, and this cannot be done without surgical excision.
Diagnostic features of FC essentially include capsular invasion, vascular invasion, and metastasis. Usually the follicular epiethelium is very benign-appearing. FAs have well-rounded, dark nuclei with abundant cytoplasm. Lesions can be microfollicular or macrofollicular and may have marked variation in cell size with focal crowding. Compared with the diagnostic features described above for PTC, these benign findings in follicular neoplasms might have otherwise raised alarm. The only way to distinguish FA from FC in lesions with no obvious extrathyroidal extension, vascular invasion, or metastasis is to microscopically visualize the entire lesional capsule to evaluate for capsular invasion, and often take many serial levels through suspicious tissue blocks. Benign adenomatous nodules will not have a capsule but usually maintain a circumscribed architecture. Adenomatous clusters of cells present within the thyroid with infiltrative borders should be scrutinized to rule out that these are metastases from an FC located elsewhere in the gland.
FC represents approximately 25% of thyroid malignancies. Somewhat more common in women, it tends to occur in middle age, with a clinical presentation similar to that of PTC. There are two subtypes of FC, minimally invasive and widely invasive. Minimally invasive carcinoma may often have no overt features of malignancy other than the demonstration of capsular invasion discussed above. Although surgery is usually curative and assertions of “overcalling” of FA as FC (minimally invasive) exist, the propensity for these neoplasms to recur 10–20 years following primary excision warrants regular clinical follow-up. Less common are widely invasive FCs, with a nastier clinical course, often presenting with extensive gross and microscopic disease. As discussed for PTC, FC may respond to radioactive iodine ablation, as a surgical adjuvant or primary treatment in advanced disease. FCs that are oncocytic (former Hürthle cell) in type (large, oxyntic cells on hematoxylin and eosin, and green and gritty on cytologic Papanicolaou stain) are often refractory to iodine treatment. As a result, although the 10-year survival rate for FC is 85%, it is less for oncocytic (Hürthle cell) carcinoma at 75%.
Wider dissemination usually occurs via the hematogenous route, in contrast to the lymphatic spread of PTC. FCs are insidious, as previously mentioned, and bone, lung, and other visceral metastases may not become evident for years.
Poorly Differentiated Thyroid Carcinoma
This entity is somewhat controversial. It is thought to be intermediate prognostically between follicular/papillary carcinomas and undifferentiated carcinomas. These tumors can often be found in association with more well differentiated carcinomas, and conversely, with undifferentiated carcinomas, and they are thought to be part of a spectrum of disease. They are most striking in their insular, solid, or trabecular growth pattern. Necrosis and increased mitoses are also present. A recent consensus conference by a group of experts sought to put together a diagnostic algorithm, but the entity is still evolving.
Undifferentiated (Anaplastic) Carcinoma
Constituting less than 5% of thyroid malignancies, undifferentiated (anaplastic) carcinomas are clinically quite distinct from papillary and follicular carcinomas. They usually present at an older age and are marked by rapid tumor growth and extensive local invasion (often with tracheal stenosis). They may present as large, stony hard masses with little mobility and often a hoarse voice due to recurrent laryngeal nerve palsy. Widespread metastatic disease is a typical finding. They are usually fatal within a year of diagnosis and may originate within long-standing, and neglected, papillary or follicular carcinomas.
Diagnosis of anaplastic carcinoma is often difficult and may require clinical and radiologic correlation. These tumors are histologically diverse, with pleomorphic, spindle cell, and even paucicellular growth patterns. Additionally, the term undifferentiated stems from the fact that these tumors often lack not only the histology and appearance of thyroid cells but more importantly, in current laboratories they lack the immunophenotypic markers of thyroid differentiation (thyroglobulin and thyroid transcription factor 1) often used to distinguish conventional thyroid carcinomas. Indeed, there may be a continuum of molecular derangements that lead to their development, involving not only BRAF and RET but further hits in p53 and beta catenin.
Because these tumors are so locally aggressive and destructive and because they are often diagnosed at an advanced stage, conventional treatment modalities, surgery, and radioiodine ablation are generally ineffective. Most treatment centers have used these methods, along with radiation and traditional chemotherapy, largely in a palliative role. As mentioned, patients with this diagnosis largely succumb to disease within weeks to months, with rare patients living for a couple of years. Longer survival is probably associated with misdiagnosis.
Medullary Carcinoma
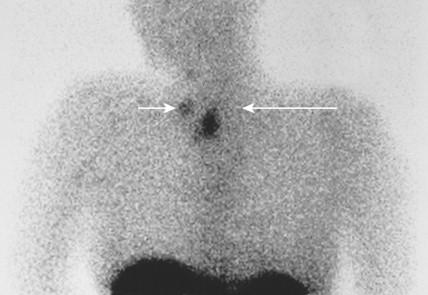
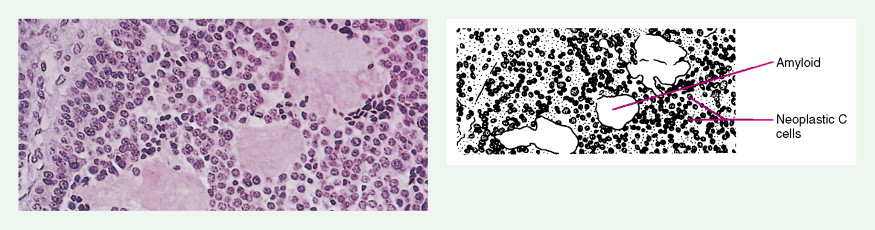
Medullary thyroid carcinomas (MTCs) arise from parafollicular C cells and account for 5% to 10% of thyroid malignancies. C cells are derived from the neural crest and secrete calcitonin, as well as other polypeptides, such as vasoactive intestinal polypeptide (VIP), carcinoembryonic antigen (CEA), somatostatin, and corticotropin (ACTH). Tumor cells specifically stain with antibodies directed against calcitonin and CEA, but may also show reactivity for neuroendocrine markers such as neuron-specific enolase, synaptophysin, and chromogranin.
MTC usually presents as a solitary solid mass, often with encapsulation. Vascular invasion is common, as is involvement of regional lymph nodes. Severe watery diarrhea, an effect of VIP secretion, is present in some patients. The majority of cases are sporadic and occur in patients over the age of 40; the disease is usually unilateral. This malignancy also occurs as part of the MEN II syndrome, in which case the incidence involves a younger age group and is uniformly bilateral. As a result of routine screening of family members for mutations within the RET oncogene and for elevated calcitonin levels, a progressively younger age group with smaller tumors or even preclinical C-cell hyperplasia is now being identified. A non-MEN familial type also exists; it is characteristically bilateral.
STAGING OF THYROID CANCER
The primary determinants of prognosis in thyroid cancer are tumor factors (size, local invasion, lymph node involvement, histology) and host factors (age and gender). In the staging system for thyroid cancers, stage I is marked by confinement of the tumor to the thyroid gland; involvement may be unilateral, bilateral, or multifocal. With stage II disease the tumor is localized to the gland, with movable regional lymph nodes. Direct local invasion or fixed regional nodes mark stage III tumors, and the presence of distant metastases characterizes stage IV lesions.
Additional factors are taken into consideration in determining prognosis. With PTC, presentation at an older age (generally >50 years), tumor size and extent (extrathyroidal extension), cytologic atypia, and lack of differentiation are all unfavorable prognostic factors. FCs that are minimally invasive carry a favorable prognosis. Extension beyond the thyroid, tumor size, and an older age at presentation are likewise unfavorable prognostic features in FC, together with nodal involvement and spread to distant sites. In medullary carcinoma, patients with the sporadic form of the disease usually have a poorer prognosis than those with the familial form, perhaps because of the earlier diagnosis of the latter. Other unfavorable prognostic factors include metastatic disease, tumor size, an older age at diagnosis, male sex, the presence of diarrhea, and rapidly increasing calcitonin levels.
CLINICAL MANIFESTATIONS
Most patients (75%) present with a neck mass. Uncommon manifestations include rapid thyroid enlargement, neck pain, dysphagia, hoarseness, and hyper- or hypothyroidism. The diagnostic approach to the incidentally discovered thyroid nodule is undergoing constant change. Because benign nodules greatly outnumber thyroid cancers, accurate diagnosis is imperative to avoid unnecessary surgery. With the exception of an elevated calcitonin level in medullary carcinoma, the only definitive diagnostic test is biopsy by needle aspiration. Lesions suspicious or diagnostic for malignancy can then be surgically evaluated.
Thyroid scanning is uncertain, because hypofunction is common in benign nodules and only 6% to 20% of “cold” nodules are malignant. Conversely, if a nodule is hyperfunctional (i.e., “hot”) on thyroid scan, the likelihood of malignancy is very low but not absent. Because a cystic lesion is less likely to be malignant, ultrasonography has been used to distinguish benign from malignant nodules. Cystic lesions, however, represent only 10% to 25% of thyroid nodules. The cystic nature of the lesion must be confirmed by aspiration and cytologic examination. Because most cysts arise from spontaneous bleeding into small, solid lesions, even a negative cytologic examination does not erase the possibility that a cyst might have been caused by a subcentimeter carcinoma. The prevalence of cancer within a cyst ranges from 0.6% to 2%, the same incidence as in solid nodules overall.
MOLECULAR BIOLOGY
Determination of DNA ploidy status by flow cytometry or growth activity by Ki-67 is not of value in discriminating between FAs and well-differentiated FCs. About 10% to 20% of PTCs are associated with activation of an oncogene named papillary thyroid carcinoma (PTC). PTC is derived from a rearrangement of the RET proto-oncogene mapped to chromosome 10q11.2. Sequential dedifferentiation of FCs is associated with overexpression of TP53 gene mutations and beta catenin mutations (as may be found in poorly differentiated and undifferentiated thyroid cancers). In addition, RAS genes are frequently mutated in these tumors. Mutations of mitochondrial DNA are reported in PTC. Some FCs of the thyroid may contain a PAX-PPAR γ chromosomal translocation, not seen with PTCs or with FAs, although this is not a consistent finding. Patients with familial MTC would be expected to possess a mutation in the RET proto-oncogene as is seen in MEN II. These defects occur much less frequently in non-MEN sporadic MTC.
TUMORS OF THE PARATHYROID GLANDS
Parathyroid tumors are most commonly benign and can be identified on the basis of the biochemical abnormality that they induce: hypercalcemia. Parathyroid tissue expresses extracellular calcium receptors. Parathyroid tumors are characterized by deficient expression of these receptors, resulting in impaired calcium regulation. In effect, the tumor senses that the serum ionized calcium concentration is lower than in reality, causing the tumor to secrete parathyroid hormone (PTH) at a physiologically inappropriately elevated level. High levels of PTH unresponsive to physiologic inihibitors result in hypercalcemia, bone demineralization, osteopenia (acutely, osteoporosis if chronic), hypercalciuria, and nephrolithiasis.
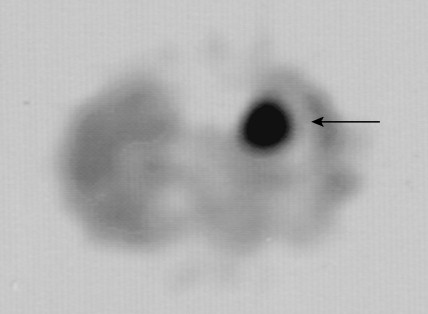
The most common parathyroid neoplasm is a solitary adenoma. Adenomas can be located anywhere from the angle of the jaw to the lower mediastinum. However, more than 99% are within the neck and can be identified based on mitochondrial uptake of radiolabeled sestamibi or on gross morphology alone. These cause an indolent disease extending over 5 or more years before detection.
In contrast to parathyroid adenomas, parathyroid carcinomas may cause rapidly progressive hypercalcemia with higher calcium and PTH concentrations than are seen customarily with the diagnosis of hyperparathyroidism. Histologically, these tumors are characterized by large size (>5 g), broad fibrous septae, increased mitotic rate as shown by immunostaining with the monoclonal antibody MIB-1 (MIB-1 proliferative index >10%), vascular invasion, extraparathyroidal extension, and most definitively by metastasis. These carcinomas are quite rare and represent 1% to 2% of cases of hyperparathyroidism. Clinically, they often present with serum PTH levels from hundreds to thousands of picograms per milliliter. Although these tumors are usually hard and fibrotic as compared with the softer nodules of adenoma, grossly, long-standing parathyroid adenomas can become fibrotic with degenerative atypia, and care should be taken before making a diagnosis of carcinoma. The only true marker of malignancy in parathyroid glands is metastasis. However, with several of the above-mentioned features (size, vascular invasion, mitotic index, etc.), the possibility should be communicated for clinical follow-up. Additionally, there have also been non–hormone-producing parathyroid carcinomas, although this is quite rare.
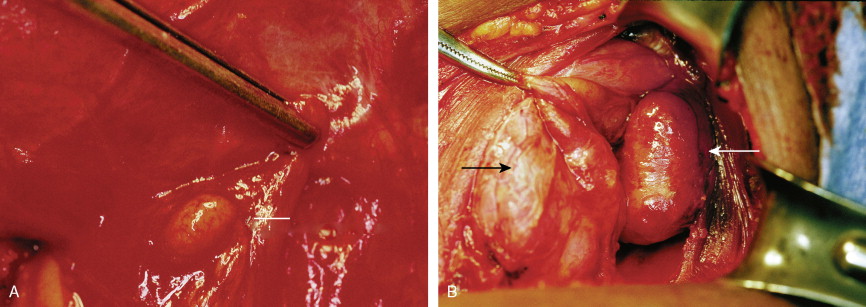
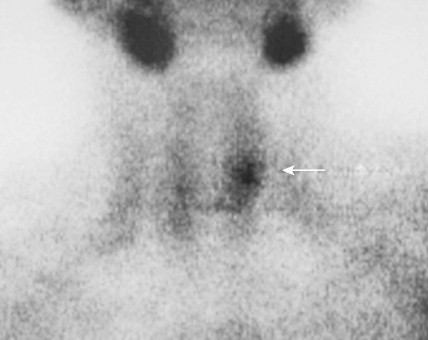
Tumors of the Adrenal Gland
The adrenal gland is composed of two defined areas, the adrenal cortex and the adrenal medulla. The outer cortex contains the steroid hormones, glucocorticoids, and mineralocorticoids. Benign tumors of the adrenal cortex are adenomas. These may be discovered incidentally or secondary to hypersecretion of feedback-insensitive hormone-producing cells. Hypersecretion can result in clinical manifestations of Cushing syndrome, Conn syndrome, aldosteronism, and feminization. Although these adenomas have no metastatic potential, their hormone secretion can result in life-threatening metabolic aberrations. Hypersecretion of adrenal cortical hormones may result from a solitary adenoma, multiple adenomas, or nodular hyperplasia, depending upon whether or not this aberration is a result of a primary process or is occurring secondarily as a result of an outside stimulus ( Table 11.2 ).
| Major Steroid Secreted | Clinical Manifestations |
|---|---|
| Glucocorticoid |
|
| Mineralocorticoid | Conn syndrome (hypertension and hypokalemic alkalosis) resulting from primary hyperaldosteronism |
| Androgen |
|
| Estrogen |
|
The incidence of adrenocortical tumors is poorly defined. However, autopsy series suggest that asymptomatic adrenal adenomas are present in 2% of adult patients, in approximately 30% of elderly, obese diabetic patients, and in up to 20% of hypertensive patients. Computed tomography (CT) scans for other conditions identify unsuspected “incidental” adenomas at the same rate. In familial MEN syndromes the autopsy incidence of adrenal adenoma seems to be 33%. About 20% of patients with Cushing syndrome have adrenal tumors, benign tumors being slightly more common than malignant tumors in the adult. An estimated 130 new cases of adrenocortical carcinoma are diagnosed each year in the United States, with the peak incidence occurring in the fourth and fifth decades.
MORPHOLOGY
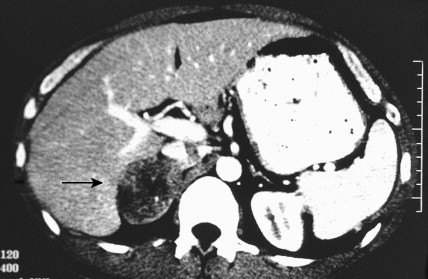
Grossly benign cortical adenomas are often bright yellow or orange, are well-circumscribed, and range from a couple of millimeters in size to several centimeters. Due to rapid or long-standing growth, adenomas may have variable amounts of hemorrhage, fibrosis, calcification, and/or cystic degeneration. Adrenocortical carcinomas are usually discovered quite late in their clinical course and are usually quite large, often larger than 10 cm in greatest dimension and weighing over 100 g. The reasons for this are that the majority of them do not produce exogenous adrenocortical hormones and therefore do not cause symptoms, growing quite large in the potential space of the retroperitoneum, becoming symptomatic as they invade vessels, metastasize, and become problematic due to mass effect. They are usually larger than 100 g and are often metastatic when diagnosed; they typically show a considerable degree of hemorrhage, necrosis, and calcification. The distinction between benign and malignant tumors is difficult to make on the basis of morphologic characteristics. However, vascular or capsular invasion and distant metastases are certain signs of malignancy, as is gross local invasion at the time of surgical resection. Although extensive necrosis, hemorrhage, numerous mitoses, and cellular and nuclear pleomorphism are more likely to be seen in malignant tumors, these features are not reliable indicators of malignancy.
CLINICAL MANIFESTATIONS
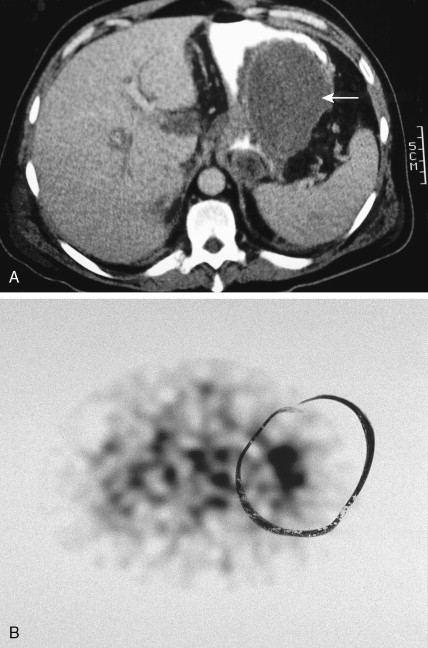
Tumors of the adrenal cortex may be functional or nonfunctional in their ability to synthesize and secrete biologically active steroid molecules. Patients with nonfunctional tumors typically present with manifestations attributable to a large abdominal mass, which is palpable in less than 50% of cases. The most common presentation is on a CT scan for persistent back pain. This is typical of adrenocortical carcinomas, which are inefficient producers of steroids. Patients with functioning tumors present with features attributable to the predominant excess steroid produced. The hypothalamic-pituitary-adrenal axis and the effects of its interaction with exogenous or endogenous steroids or ACTH are shown in Figure 11.27 . Also depicted are the effects on this axis of primary pituitary and adrenal tumors, as well as ectopic tumors.
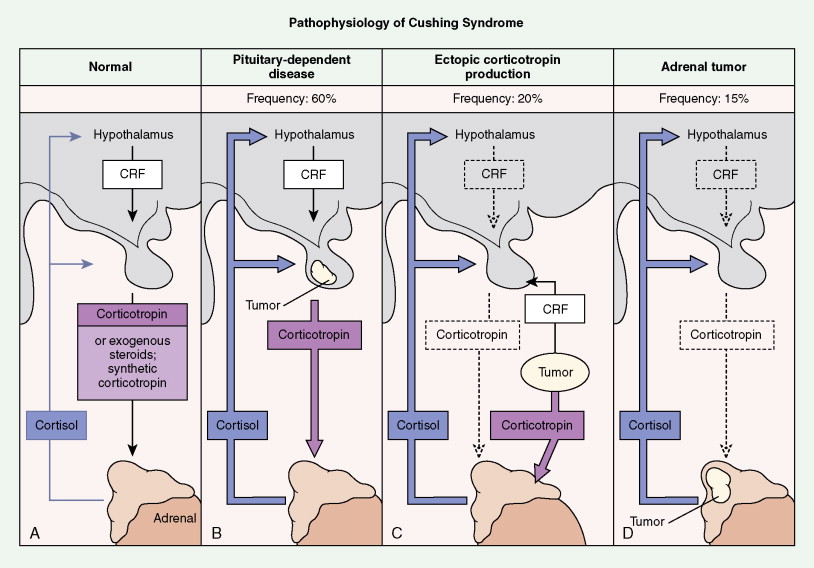
Although in most cases Cushing syndrome has either a primary pituitary or an adrenal etiology, ectopic elaboration of ACTH is also an important cause (see Fig. 11.27 ). Ectopic secretion of ACTH is usually associated with small cell lung cancer, although other tumors reported to secrete ectopic ACTH include carcinoid tumor, pheochromocytoma, MTC, thymoma, and even pleural-based mesotheliomas. Excess mineralocorticoid secretion is usually caused by benign cortical adenomas and is only rarely observed in association with adrenocortical carcinoma. Syndromes of virilization or feminization and precocious puberty are also seen in primary adrenal enzymatic disorders, as well as in primary hypothalamic, gonadal disorders and adrenal tumors.
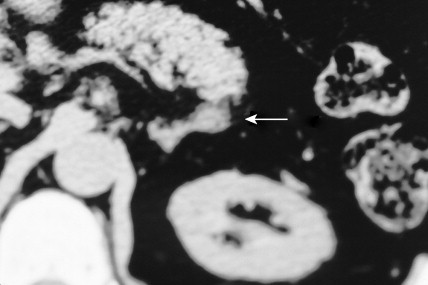
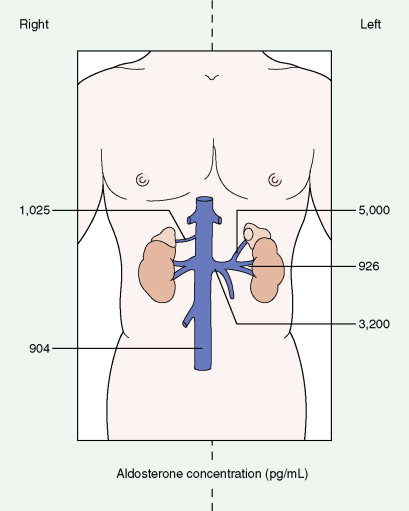
Once the diagnosis of excess adrenocortical hormone secretion has been made, attempts at localization are undertaken. CT scanning is useful for localizing adrenal tumors and can exclude local extension from tumors of other abdominal organs. Adrenal hyperplasia is marked by bilateral enlargement, which is usually symmetrical, whereas adrenal tumors are usually unilateral and can be quite massive. Examination of the inferior vena cava by CT scan can document venous invasion in some malignant tumors. Intravenous pyelography and ultrasonography have largely been supplanted by CT. Angiography can be used to map the arterial supply of a tumor and may reveal hepatic metastases. Adrenal venous sampling, often used when radiographic evaluation cannot localize the primary tumor, can accurately localize almost all aldosteronomas.
The most common primary tumors metastatic to the adrenals are lung, breast, gastric, and colorectal tumors and melanoma. Although the involvement is usually unilateral, bilateral metastases sometimes occur. Adrenal insufficiency is uncommon and should be ruled out by an ACTH stimulation test. CT-guided fine-needle aspiration is of value in distinguishing between primary and metastatic tumors.
MOLECULAR BIOLOGY
Establishing a diagnosis of malignancy in adrenocortical tumors is best performed based upon clinical pathologic criteria. Molecular markers are not yet of use in the diagnosis or prognosis of adrenocortical tumors.

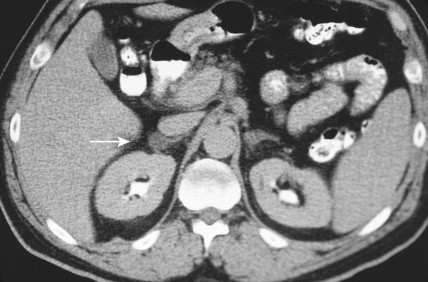
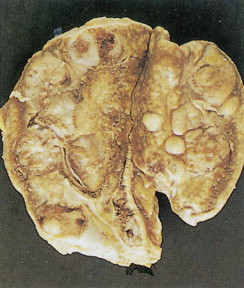
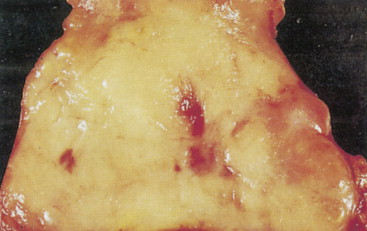
Pheochromocytoma
Pheochromocytoma is a neoplasm of the chromaffin cells of the sympathoadrenal system. It is, by definition, a paraganglioma of the adrenal medulla (see below). It occurs in 0.1% to 1% of hypertensive individuals, with an incidence rate of 0.95 per 100,000 person-years. The peak incidence occurs in the third to fifth decades, without a sex predilection. Although it is typically associated with only a 10% familial etiology, we are increasingly recognizing the genetic linkages in these patients, with identifiable mutations in up to 30% of cases. Many of the familial cases are inherited as an autosomal-dominant trait, either independently or as part of MEN II syndrome (in which case the neoplasm is bilateral in 70% of patients) or familial paragangliomatosis, with mutated SDH (succinate dehydrogenase) genes. Associated heritable disorders include von Recklinghausen’s neurofibromatosis, von Hippel-Lindau syndrome, and cerebellar hemangioblastoma.
Pheochromocytomas arise within the adrenal medulla in 90% of cases. Extra-adrenal paragangliomas usually occur intra-abdominally within the lower para-aortic sympathetic chains, also called the organs of Zuckerkandl. The tumors range in size from a few grams to 3 kg and are usually encapsulated, highly vascular, and yellowish to reddish brown in color. Hemorrhage or necrosis, or both, is often present. Microscopically, the tumor is composed of clusters of finely granular and basophilic or eosinophilic cells. They are grouped into organized clusters of cells, “zellballen,” and they are surrounded by rare, spindled sustentacular cells (positive for S-100 protein by immunohistochemistry). The lesional cells stain for chromogranin and synaptophysin, and in difficult cases, may be distinguished from adrenocortical tumors by negativity for inhibin. In patients with neurofibromatosis a composite tumor may be found, composed of a mixture of pheochromocytoma and ganglioneuroma.
Electron microscopy identifies neuroendocrine granules containing norepinephrine or epinephrine. Only 5% to 10% of cases are malignant, and malignancy can be confirmed only by the presence of direct extension into surrounding structures or distant metastases.
The diagnosis of pheochromocytoma is confirmed by the demonstration of excessive levels of catecholamines or their metabolites in urine or plasma. Measurements of 24-hour urinary catecholamines and their metabolites, vanillylmandelic acid and metanephrines, remain the standard diagnostic tests. Metanephrine measurements are the most sensitive and are considered the best method of screening. One or more of these measurements are positive in 95% of cases. Pharmacologic tests, either provocative or suppressive, are rarely needed to make the diagnosis.
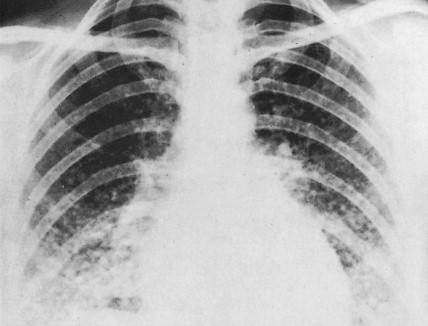
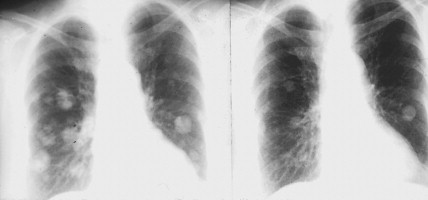
Localization of pheochromocytomas is most often accomplished noninvasively by use of CT or ultrasonography. Abdominal CT is positive in more than 90% of cases and allows localization of extra-adrenal pheochromocytomas. Chest radiographs may detect primary intrathoracic tumors or metastases from a malignant pheochromocytoma. [ 131 I]metaiodobenzyl guanidine (MIBG) scintigraphy has been very useful in localizing tumors. IMBG, an analogue of guanethidine, is taken up by adrenergic storage vesicles. Most pheochromocytomas express receptors for the hormone somatostatin. Thus, imaging with radiolabeled somatostatin analogue (octreotide) has also been useful.
The clinical manifestations of pheochromocytoma are attributable to increased release of catecholamines, mainly epinephrine. Hypertension, either sustained or episodic, is present in the majority of patients, often associated with headache, palpitations, excessive sweating, tremors, nausea/vomiting, and flushing. Hypotension can be seen in some epinephrine-secreting tumors. A normal blood pressure may be associated with dopamine-secreting pheochromocytomas, which are mostly extra-adrenal. Other symptoms include orthostatic hypotension, atrial and ventricular arrhythmias, impaired glucose tolerance, constipation, and symptoms of hypermetabolism such as heat intolerance and weight loss. In several studies, however, a significant proportion (15% to 20%) of pheochromocytomas were clinically unsuspected and diagnosed at autopsy.
Paraganglioma
The paraganglia consist of widely dispersed collections of the neural crest cells that arise in association with segmental or collateral autonomic ganglia throughout the body. The extra-adrenal paraganglionic system is divided into four anatomic groups. Tumors that arise in the branchiomeric or intravagal paraganglia are typically chromaffin-negative and are therefore nonfunctional, whereas tumors in the aorticosympathetic paraganglia have variable chromaffin affinity and functional activity. Viscero-autonomic paraganglia are associated with viscera such as the urinary bladder, gallbladder, and intrathoracic structures.
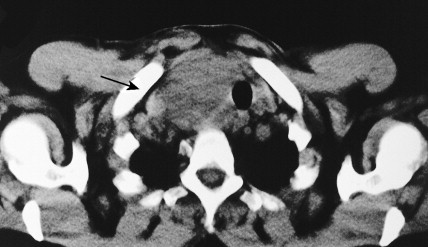
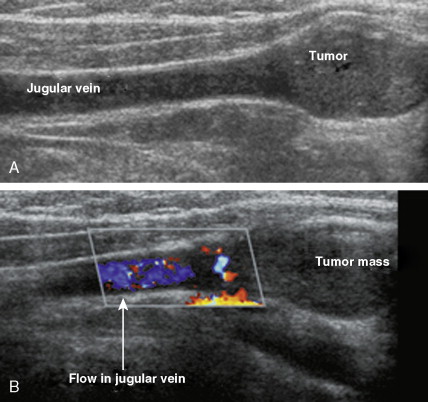
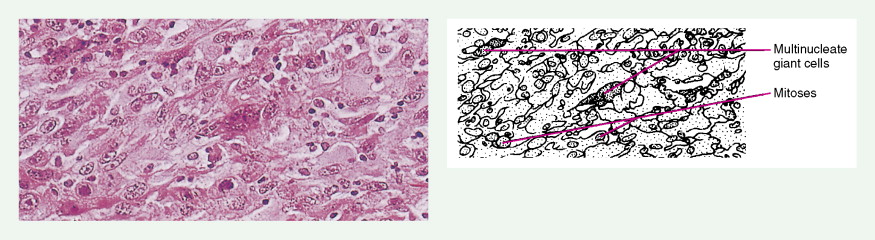
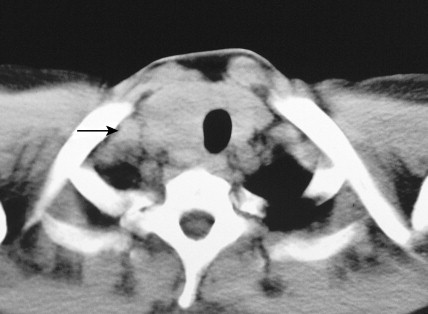
Carotid body tumors (chemodectoma, non-chromaffin paraganglioma) are the most common of the extra-adrenal paragangliomas. They are highly vascular lesions, usually arising from and adherent to the bifurcation of the common carotid artery. They are inherited as an autosomal-dominant trait. Patients typically present between the ages of 40 and 60 years with a painless, slowly enlarging mass in the upper neck below the angle of the jaw; the mass is bilateral in 2% to 5% of cases. The tumors are rarely functional and must be distinguished from an extra-adrenal paraganglioma arising in the cervical sympathetic chain. Although carotid body tumors are usually benign, in up to 10% of cases they may behave in a malignant fashion, invading locally.
Paragangliomas involving the temporal bone and middle ear (glomus jugulare tumors) constitute the second most common extra-adrenal paragangliomas. They usually occur in middle-aged women, who present with dizziness, tinnitus, and conductive hearing loss. Cranial nerve palsies, seen in 40% of patients, result from tumor extension to the base of the brain. Less than 1% of the tumors are functional.
Vagal paragangliomas (vagal body or glomus vagale tumors) usually arise between the mastoid process and the angle of the jaw in the parapharyngeal space. Patients typically present with necrologic symptoms secondary to cranial nerve palsies (tongue weakness, vocal cord paralysis, hoarseness, and Horner syndrome). Vagal body tumors are more common in women and are multiple in 10% to 15% of cases. They are rarely functional and are often locally invasive, with lymph node metastases.
Mediastinal paragangliomas usually present as asymptomatic anterior or superior mediastinal masses. Posterior mediastinal paragangliomas are less common, typically occur in younger patients, and are more often associated with functional activity. Metastases are seen in up to 10% of patients and locally aggressive disease in 20% to 30%.
Retroperitoneal paragangliomas, which typically arise adjacent to the adrenal glands, manifest in a younger age group (30–40 years) than head and neck paragangliomas. These can also occur at the aortic bifurcation in the organs of Zuckerkandl. Back pain and a palpable mass are the two most common symptoms, although 10% of patients present with metastatic disease. Functional symptoms caused by production of norepinephrine occur in 25% to 60% of patients. The tumors are typically large and chromaffin-positive, and they behave more aggressively than their adrenal counterparts. Metastases occur in 20% to 40% of patients, as compared with only 2% to 10% of patients with adrenal pheochromocytomas.
MOLECULAR BIOLOGY
The importance of molecular studies in paragangliomas is mostly in the context of familial syndromes. There have been no markers of malignancy that have been helpful thus far, although panels, including such markers as stathmin, found to be overexpressed in malignant neoplasms as well as paragangliomas, may be useful. Patients with familial syndromes, particularly with mutations in the SDHB gene, are more likely to have malignant disease. Immunohistochemical markers are commercially available to screen excised tumors for changes in SDH gene expression. If there is a suspected altered gene locus, either by family history or by tumor expression profile, further genetic workup should be considered—particularly for MEN syndromes, von Hippel Lindau syndrome, or familial paragangliomatosis, for which molecular characterization is more established.