Visit the Endocrine Hypertension: From Basic Science to Clinical Practice , First Edition companion web site at: https://www.elsevier.com/books-and-journals/book-companion/9780323961202 .




Introduction
Hypertension (HTN) is a significant cause of cardiovascular and cerebrovascular morbidity and mortality in both children and adults. The prevalence and etiology of secondary HTN vary by age [ ]. Approximately 5%–10% of adults and 75%–85% of children with HTN may have secondary HTN with an underlying and potentially curable cause.
Most of the monogenic causes of endocrine HTN resulting from single-gene mutations such as Liddle’s syndrome, congenital adrenal hyperplasia (CAH) subtypes, apparent mineralocorticoid excess (AME), familial hyperaldosteronism (FH) subtypes including glucocorticoid-remediable aldosteronism (GRA), and familial primary generalized glucocorticoid resistance (Chrousos syndrome) increase the reabsorption of sodium in exchange with potassium leading to hypokalemic alkalosis, volume expansion and HTN with low plasma renin activity (PRA) [ ]. Pseudo-hypoaldosteronism type II (Gordon syndrome) is the only monogenic HTN that manifests with hyperkalemia, metabolic acidosis, suppressed PRA and HTN due to increased sodium chloride reabsorption in the distal nephron and loss-of-function mutations in the WNK4 and CUL3 genes [ , ].
This chapter will revisit the diagnosis and management of the known causes of monogenic HTN.
Inherited disorders of endocrine hypertension
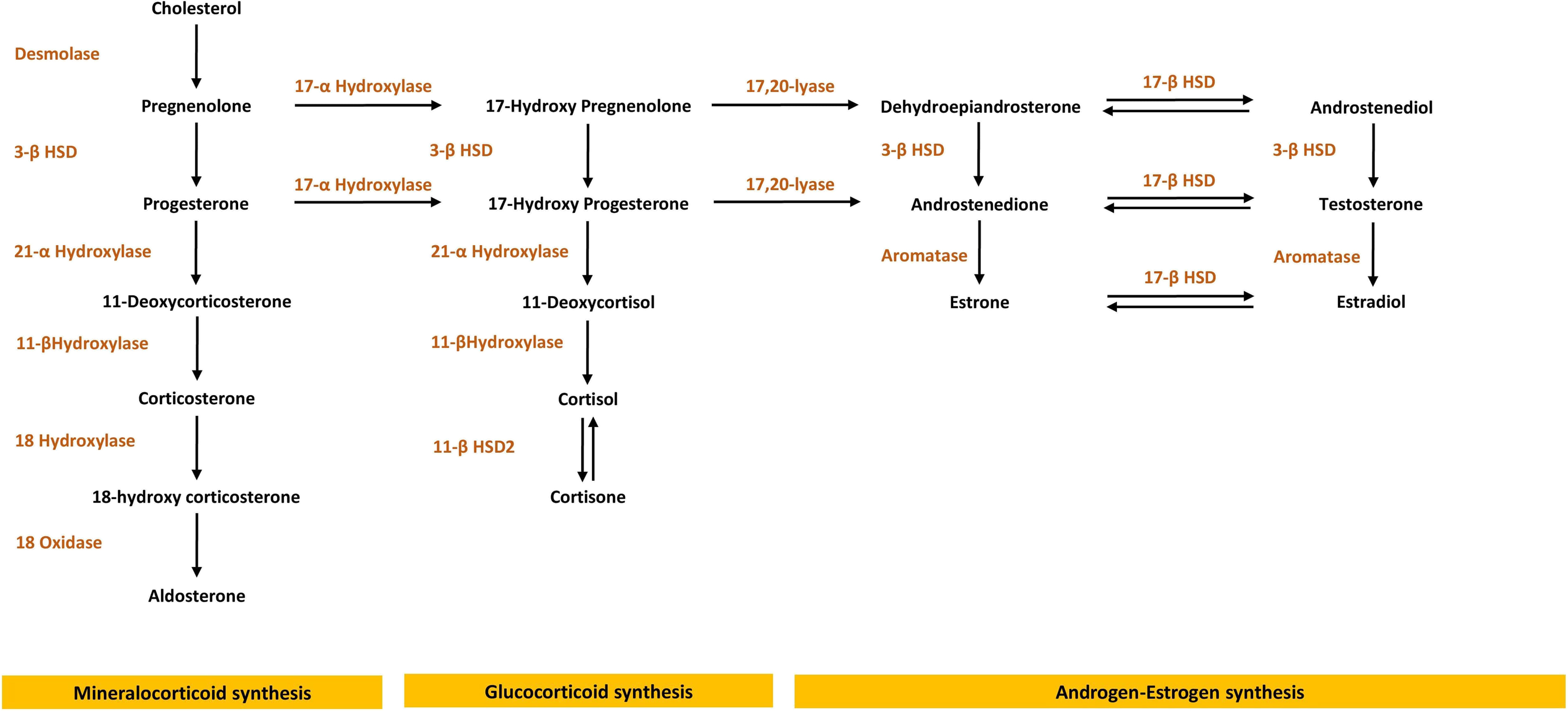
Under physiological conditions, sodium transport in the renal epithelial cells of the cortical collecting duct is regulated by the mineralocorticoid hormone aldosterone, which acts as the physiologic agonist of the mineralocorticoid receptor (MR) [ ]. Even though cortisol and aldosterone have a nearly equal affinity toward the mineralocorticoid receptor in vitro, it is only the aldosterone that acts as the mineralocorticoid receptor agonist in vivo, despite the circulating cortisol levels being three times higher than the aldosterone levels. This is mediated by the enzyme 11β-hydroxysteroid dehydrogenase type 2 (11βHSD2) that inactivates the 11-hydroxysteroids including cortisol to their inactive keto-forms, thus protecting the mineralocorticoid receptor from activation by glucocorticoids. Excessive secretion of mineralocorticoids or abnormal sensitivity to mineralocorticoid hormones may result in volume expansion and HTN. Fig. 10.1 provides an overview of the adrenal steroid synthesis pathway.
HTN secondary to mineralocorticoid excess includes a spectrum of clinical disorders ranging from overproduction of aldosterone (primary hyperaldosteronism), overactivity of the renin-angiotensin-aldosterone system (RAAS) such as renin-secreting tumors, nonaldosterone mineralocorticoid causes including CAH, AME syndrome [ ], Liddle syndrome [ , , ], the various forms of Cushing syndrome, Pseudohypoaldosteronism type II (Gordon syndrome) [ ], GRA, glucocorticoid receptor resistance syndrome (Chrousos syndrome) [ , ], and Geller syndrome [ ].
Early-onset of HTN in a young individual resistant to standard antihypertensive medications indicates that all causes of inherited disorders of endocrine HTN have to be considered [ ]. In these disorders, HTN is usually associated with hypokalemia, metabolic alkalosis, and low PRA secondary to renal sodium retention ( Table 10.1 ) [ , , ]. Severe hypokalemia, in addition to lowering the renal tubular cell pH, can also impair distal chloride reabsorption leading to increased urine chloride excretion (>15 mEq/L), and increased bicarbonate reabsorption [ , ].
| PRA and PAC levels | Medical condition | Hypokalemic alkalosis | Hyperkalemic acidosis |
|---|---|---|---|
| Low-PRA Low-PAC | Liddle’s syndrome | + | − |
| CAH | + | − | |
| AME | + | − | |
| Geller syndrome | + | − | |
| Chrousos syndrome | + | − | |
| Carney syndrome | + | − | |
| Low-PRA High-PAC | Primary aldosteronism | + | − |
| FH-1 to FH-4 | + | − | |
| High-PRA High-PAC | JGCT | + | − |
| Normal-PRA Normal-PAC | Pheochromocytoma | − | − |
| Paragangliomas | − | − | |
| Low-PRA Normal-PAC | Gordon syndrome | − | + |
Genetic forms of endocrine HTN result from activating or inactivating mutations within the mineralocorticoid, glucocorticoid, or sympathetic pathways. HTN results from excessive sodium reabsorption by hyperactive channels, hyperstimulation of MR due to increased glucocorticoid synthesis, or excessive mineralocorticoid synthesis.
It is helpful to approach patients with resistant HTN and hypokalemic alkalosis by obtaining plasma aldosterone concentration (PAC) and PRA as an initial step. This helps to separate patients into one of the three categories: (1) Those with suppressed PAC and PRA; (2) those with a high PAC and low PRA; (3) those with elevated PAC and PRA. Fig. 10.2 shows the diagnostic evaluation for monogenic HTN associated with hypokalemic alkalosis.
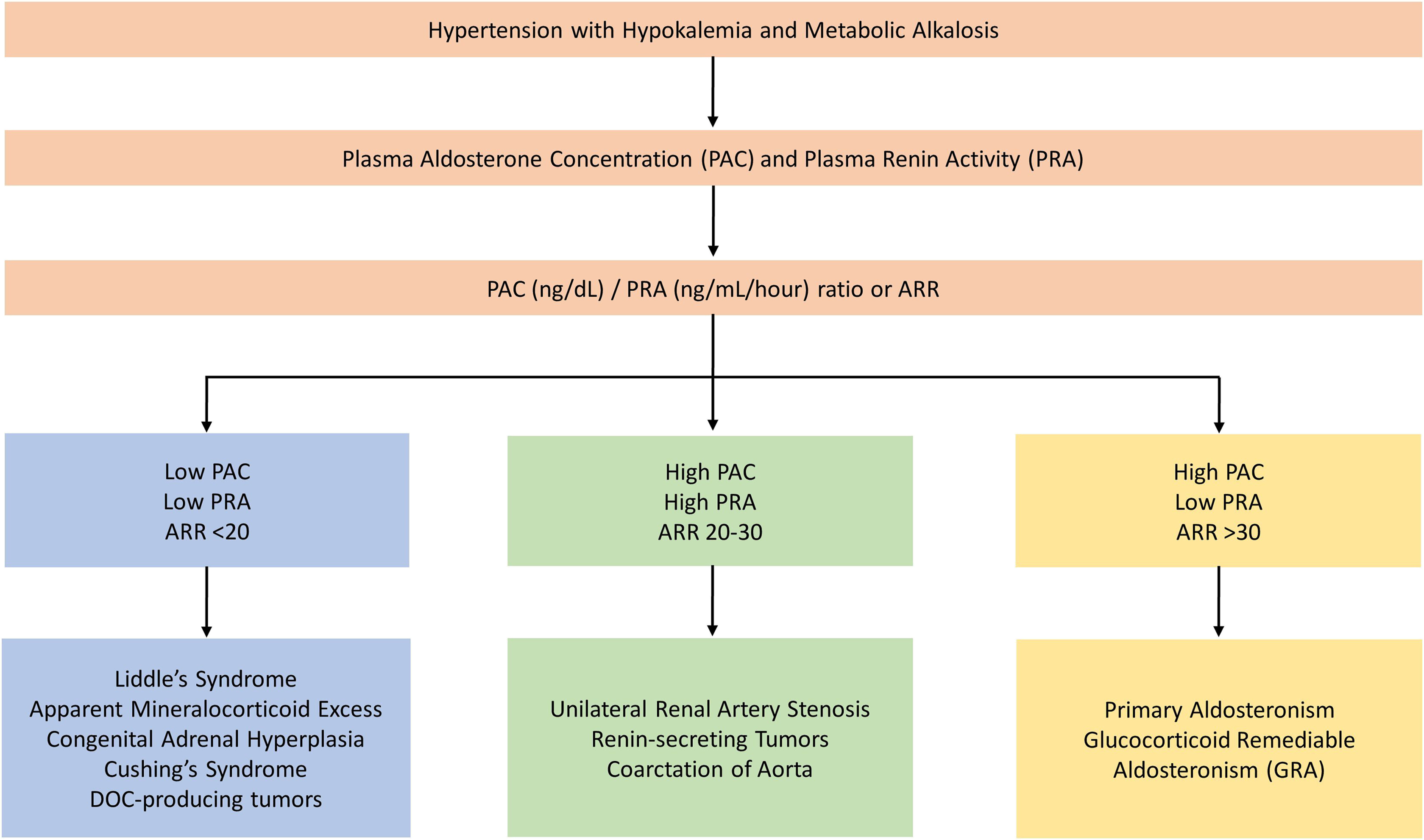
The ratio of PAC to PRA (Aldosterone renin ratio; ARR) is considered to be the best initial screening test for differentiating primary from secondary causes of hyperaldosteronism [ ]. An ARR of at least 35 has 100% sensitivity and 92.3% specificity in diagnosing primary aldosteronism when baseline plasma aldosterone level is elevated above 550 pmol/L (20 ng/mL). However, normal PAC and ARR values have been reported in hypertensive patients with primary hyperaldosteronism [ ], Thus, in hypertensive patients with normal PAC and high ARR, dynamic confirmatory testing is required to exclude false-positive results. Confirmatory tests include aldosterone suppression by saline loading, fludrocortisone administration, or converting enzyme inhibition by captopril or renin stimulation by furosemide administration [ ].
The ARR should be measured in the context of an unrestricted sodium intake and values obtained in the upright position are more sensitive than supine test results.
Monogenic hypertension associated with suppressed PAC and PRA
Liddle syndrome
Epidemiology
The disease prevalence has not yet been established. Less than 80 patients have been formally diagnosed worldwide with an estimated prevalence of 1.5% documented with genetic testing among patients with early-onset HTN in a study of the Chinese population [ ]. One study from the US among hypertensives with hypokalemia or high serum bicarbonate has shown that there was approximately a 6% prevalence of symptomatic Liddle syndrome.
The disease can be clinically heterogeneous, ranging from mild to severe cases. A lack of family history of early-onset HTN does not preclude the diagnosis of Liddle syndrome [ , ].
Pathophysiology
Liddle syndrome has an autosomal dominant inheritance and is caused by a mutation that increases the activity of the epithelial sodium channel (ENaC) in the aldosterone-sensitive cortical collecting duct that increases the sodium reabsorption [ , , , ]. The increased sodium reabsorption in this syndrome is independent of aldosterone and is the cause of suppressed PAC and PRA. HTN is associated with hypokalemic alkalosis, as the increased sodium reabsorption increases the net negative potential of the collecting duct lumen, causing an increase in urinary hydrogen ion and potassium excretion.
Genetics
Liddle syndrome is associated with gain-of-function mutations in the genes SCNN1A , SCNN1B , or SCNN1G , encoding the α, β, and γ-subunits of the (ENaC) [ ] of which SCNN1B and SCNN1G are the most commonly affected [ , ]. These genes are located on different chromosomes: SCNN1A is situated on chromosome 12p13.31, SCNN1B and SCNN1G are on chromosome 16p12.2 [ ]. These mutations cause the kidney to increase sodium reabsorption in exchange for potassium excretion, independent of aldosterone regulation ( Table 10.2 ) [ , , ].
| Medical condition | Inheritance | Pathophysiology | Prevalence |
|---|---|---|---|
| Liddle syndrome | AD | Mutations in SCNN1A, SCNN1B, or SCNN1G that encode the α, β, and γ subunits of the ENaC gene result in volume expansion and hypertension | Unknown |
| CAH | AR | Mutations in CYP11B1 and CYP17A1 encoding 11β-hydroxylase and 17-α hydroxylase cause an accumulation of intermediate steroids with MR activity | 1/10,000 |
| AME | AR | Inactivating mutations in 11 βHSD2 gene that encodes 11β-hydroxysteroid dehydrogenase type 2 allows excess cortisol to activate the MR | Unknown |
| Geller syndrome | AD | Gain-of-function mutations in MR gene NR3C2 allow atypical stimulation by other steroids | Unknown |
| Gordon syndrome | AD | Mutations in WNK1,3,4, KLHL3, and CUL3 genes that modulate the thiazide-sensitive Na + -Cl – co-transporter channel (NCC) allow unchecked activity, causing volume expansion and hypertension | Unknown |
| FH-1 (GRA) | AD | Unequal crossing over between the CYP11B1 and CYP11B2 genes generates a metabolite that is ACTH sensitive but produces aldosterone | Unknown |
| FH-2 | AD | Activating mutations in CLCN2 gene that encodes a chloride channel resulting in excess aldosterone production independent of ACTH | Unknown |
| FH-3 | AD | Activating mutations in KCNJ5 gene that encodes a potassium channel, stimulating aldosterone synthase activity independent of ACTH | Unknown |
| FH-4 | AD | Activating mutations in CACNA1H gene that encodes calcium channels in the zona glomerulosa, enhancing aldosterone synthase activity independent of ACTH | Unknown |
| Familial PPGL | AD | Multiple gene mutations including RET, VHL, NF1, SDHB, SDHD, SDHAF2, SDHC, MAX, and TMEM127 produce catecholamine-secreting tumors | Unknown |
| Chrousos syndrome | AD | Inactivating mutations on NR3C1 gene that encodes the glucocorticoid receptor leads to generalized resistance to glucocorticoids in almost all tissues, excessive ACTH secretion and hypertension | Unknown |
Clinical features
The onset of HTN is typically at a young age, between late childhood and adolescence [ ]. The youngest patient reported in the literature with Liddle syndrome was a 10-week-old girl who manifested with HTN [ ]. The typical clinical presentations include HTN, resistance to conventional therapy associated with hypokalemic alkalosis, low levels of PAC and PRA, and a family history of early-onset HTN ( Table 10.1 ) [ , , ].
Investigations
Liddle syndrome must be distinguished from other forms of monogenetic endocrine HTN associated with hypokalemic alkalosis. The suppressed PAC and PRA along with an ARR less than 30 and differential response to potassium-sparing diuretics separates Liddle syndrome from primary hyperaldosteronism [ ]. A urine steroid profile and genetic testing can distinguish it from AME and GRA ( Table 10.3 ) [ ].
| Medical condition | Blood test | Urine test | Genetic test |
|---|---|---|---|
| Liddle syndrome | – | – | Available |
| CAH | Increased 11-deoxycorticosterone and 11-deoxycortisol following ACTH challenge | – | Available |
| AME | Plasma-free cortisol > plasma-free cortisone | Urinary free cortisol > free cortisone; urinary metabolites of cortisol (tetrahydrocortisol and 5α-tetrahydrocortisol) > urinary metabolites of cortisone (tetrahydrocortisone) (THF + allo-THF/THE) | Available |
| Geller syndrome | – | – | Available |
| Gordon syndrome | Normal PAC | – | Available |
| FH-1 (GRA) | – | High urine 18-oxocortisol and 18-hydroxycortisol | Available |
| FH-2 | – | Normal urine 18-oxocortisol and 18-hydroxycortisol levels | Available |
| FH-3 | – | High urine 18-oxocortisol and 18-hydroxycortisol | Available |
| PPGL | High plasma-free metanephrine levels | High urinary fractionated metanephrine levels | Available |
| Chrousos syndrome | Elevated cortisol and ACTH levels | High 24-h free cortisol excretion | Available |
| JGCT | Elevated PAC and PRA levels | – | – |
The lack of therapeutic response to spironolactone and a good response to amiloride or triamterene strongly favors the diagnosis of Liddle syndrome ( Table 10.4 ) [ , ].
| Medical condition | Response to MRA | Response to glucocorticoids | Response to BP-lowering agents |
|---|---|---|---|
| Liddle syndrome | − | − | ENaC blockers like amiloride or triamterene |
| CAH | + | + | − |
| AME | + | + | − |
| Geller syndrome | − | − | − |
| Gordon syndrome | − | − | Salt restriction/thiazide diuretics |
| FH-1 (GRA) | + | + | − |
| FH-2 to FH-4 | + | − | − |
| PPGL | − | − | Alpha and beta-adrenergic blockers |
| Chrousos syndrome | − | − | High doses of mineralocorticoid-sparing synthetic glucocorticoids |
| JGCT | − | − | Surgical resection of tumor |
| Carney syndrome | − | − | Surgical resection of tumor |
Management
Treatment is based on blocking the ENaC with the administration of potassium-sparing diuretics such as amiloride or triamterene. This results in the correction of hypokalemic alkalosis and improvement of HTN. A low sodium diet is also helpful in controlling elevated blood pressure [ , , ]. . Spironolactone is not effective as the genetically altered ENaC is independent of aldosterone regulation [ , ].
Genetic testing is available to confirm the diagnosis [ ]. With proper treatment, the risk of cardiovascular and renal complications can be minimized.
Congenital adrenal hyperplasia (CAH)
Epidemiology
The overall estimated prevalence of CAH is 1/10,000 and annual incidence ranges between 1/5000 and 1/15,000. 11β-hydroxylase and 17α-hydroxylase deficiencies are associated with HTN and account for 5%–15% of CAH cases [ ].
Pathophysiology
Both 11β-hydroxylase and 17α-hydroxylase deficiencies are inherited in an autosomal recessive fashion and are caused by loss-of-function mutations in several genes that control the cortisol and aldosterone production. CAH is characterized by adrenal insufficiency and variable degree of hyper or hypo androgenic manifestations depending on the type of enzyme deficiencies and the severity of the disease [ ].
Genetics
The most common form of CAH (90%–95%) is caused by inactivating gene mutations in the CYP21A2 located on chromosome 6 [6p21.31] (encoding 21α-hydroxylase) that controls cortisol and aldosterone production, followed by CYP11B1 on chromosome 8 [8q22] (encoding 11β-hydroxylase) and CYP17A1 on chromosome 10 [10q24.3] ( encoding 17α-hydroxylase), which regulate different steps in steroid synthesis ( Table 10.2 ). The latter two subtypes of CAH (11β-hydroxylase and 17α-hydroxylase deficiencies) are known to cause monogenic HTN [ ].
Both subtypes of CAH, 11β-hydroxylase, and 17α-hydroxylase deficiencies, cause elevation of deoxycortisol and deoxycorticosterone levels with increased activity at the MR leading to significant mineralocorticoid activity independent of aldosterone regulation.
Clinical features
11β-hydroxylase deficiency (CAH type IV) prevents the conversion of deoxycorticosterone and deoxycortisol into corticosterone and cortisol. Reduction in cortisol and corticosterone syntheses with overproduction of 11-deoxycorticosterone and 11-deoxycortisol (DOC) leads to significant mineralocorticoid activity and subsequent sodium retention, hypokalemic alkalosis, HTN, reduced PRA and PAC ( Table 10.1 ) [ , ].
Patients with 11β-hydroxylase deficiency, like other CAH subtypes, can present with disorders of sexual development. Newborn girls, due to androgen excess may present with ambiguous genitalia at birth. They have a normal uterus but abnormal vaginal development. Androgen excess can cause precocious puberty and short stature in both sexes [ ].
The 21-hydroxylase deficiency can have a severe salt-wasting form and a mild non-classic form. The non-classic form from 21-hydroxylase deficiency is often not diagnosed until adolescence when the first symptoms appear. Affected females manifest acne, menstrual irregularities, anovulation, and hirsutism [ , ]. The salt-wasting forms can lead to severe dehydration and hypotension during the first few days of life.
Investigation
Diagnosis of 21α-hydroxylase deficiency is established by measuring 17-hydroxy-progesterone levels. Genetic testing confirms the diagnosis.
Diagnosis of 11β-hydroxylase is established by high levels of 11-deoxycorticosterone, 11-deoxycortisol and androgens, mainly androstenedione and dehydroepiandrosterone (DHEA) ( Fig. 10.1 ) following adrenocorticotropic hormone (ACTH) challenging test ( Table 10.3 ) [ ]. Patients with an 11β-hydroxylase deficiency often exhibit signs of androgen excess similar to 21α-hydroxylase deficiency. HTN coupled with high levels of androgens can differentiate 11β-hydroxylase deficiencies from other causes of CAH and monogenic HTN [ , ].
Affected females are born with a degree of masculinization of their external genitalia and even ambiguous genitalia. Male patients may have premature sexual development.
17α-hydroxylase deficiency (CAH type V) causes decreased cortisol production associated with the accumulation of corticosterone and deoxycorticosterone, a mineralocorticoid precursor, leading to HTN, hypokalemia, and delayed puberty in both sexes. Because the production of cortisol is blocked so early in the pathway, there is little production of sex hormones ( Table 10.1 ).
Male patients may exhibit ambiguous external genitalia and may even have a female phenotype, while females present with primary amenorrhea [ ]. Steroid analysis following ACTH-stimulation would lead to a correct diagnosis, showing atypically elevated levels of pregnenolone and progesterone relative to 17α-pregnenolone and 17α-progesterone, respectively.
In 17α-hydroxylase deficiency, levels of androgens and estrogens are low and do not rise with ACTH challenge. A definitive diagnosis is available with genetic testing for the mutation ( Table 10.3 ) [ ].
Management
Treatment of both 11β-hydroxylase and 17α-hydroxylase deficiencies consists of administration of glucocorticoids to suppress the ACTH secretion and deoxycorticosterone synthesis, thereby inhibiting the MR stimulation. Spironolactone, amiloride, and calcium channel blockers may be further used to treat HTN [ ]. Hydrocortisone can regulate the menstrual cycle and promote fertility in adult females. Fludrocortisone is usually given as a mineralocorticoid replacement [ , , , ]. Vaginoplasty is recommended during the first year of life. Patients with a 17α-hydroxylase deficiency should be treated with glucocorticoids with the addition of sex hormone therapy when clinically indicated [ , , ].
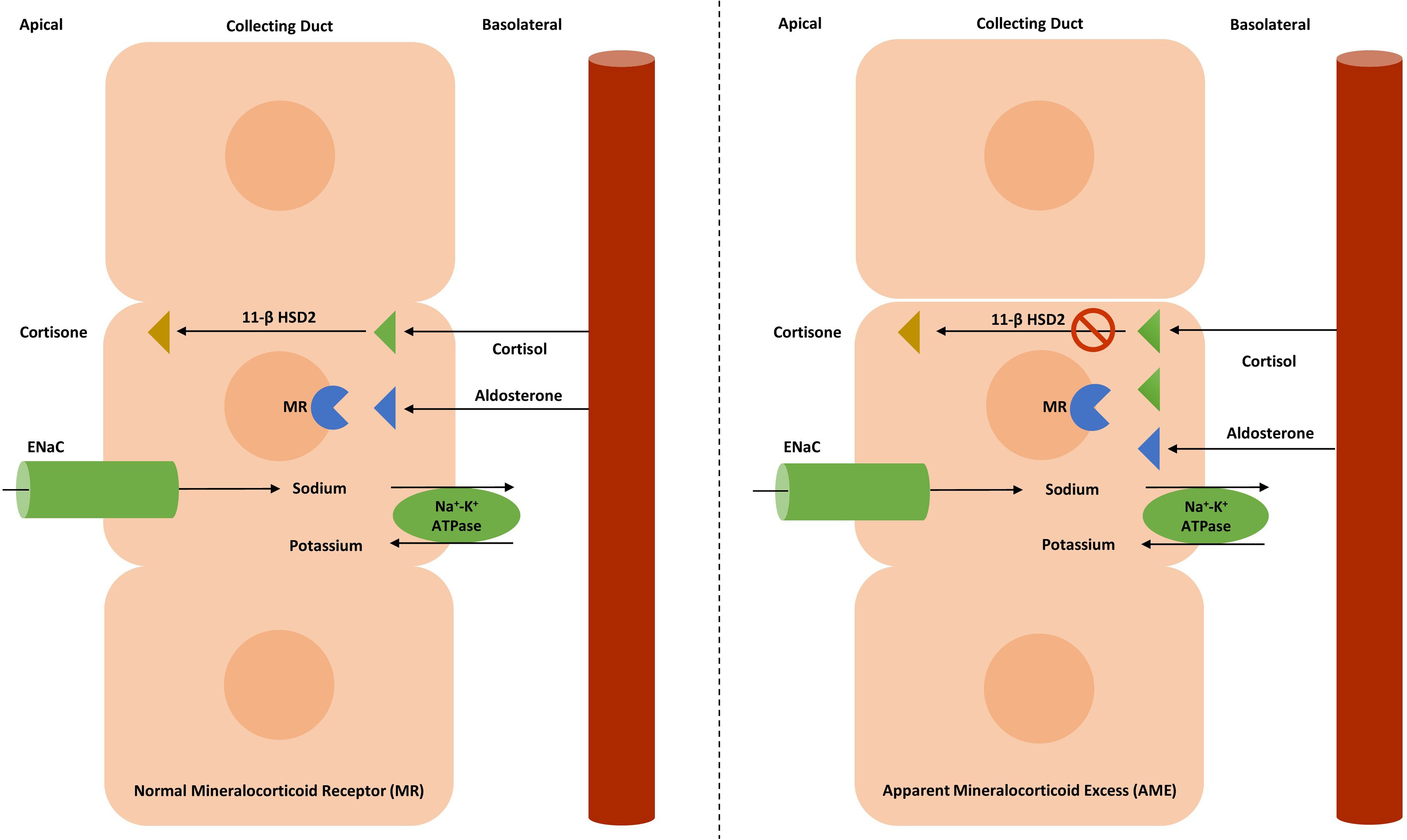
Apparent mineralocorticoid excess (AME)
Epidemiology
The prevalence varies between populations depending on the level of consanguinity. Less than 100 cases have been reported in the literature [ , ].
Pathophysiology
Normally, the isozyme 11β hydroxysteroid dehydrogenase type 2 (11βHSD2) inactivates circulating cortisol to the less-active metabolite – cortisone [ , , ]. In the absence of 11βHSD2 , excess cortisol can bind and activate the mineralocorticoid receptors in the aldosterone-sensitive principal cells of the cortical collecting duct due to nonselectivity of the receptor. However, the defect in the AME, unlike primary aldosteronism, is at the level of the MR itself rather than downstream at the ENaC channel. Unregulated mineralocorticoid receptor activation results in aldosterone-like effects in the kidney causing hypernatremia, hypokalemia, volume expansion, and HTN ( Fig. 10.3 ) [ , ].