Visit the Endocrine Hypertension: From Basic Science to Clinical Practice , First Edition companion web site at: https://www.elsevier.com/books-and-journals/book-companion/9780323961202
.


Introduction
Hypertension is a major risk factor for cardiovascular disease (CVD) and chronic kidney disease among the global population. The disease affects about 45% of the US population and significantly increases the risk of chronic morbidity and mortality among the sufferers [ ]. Similar prevalence figures are likely to exist in the other regions of the world also owing to the rising prevalence of sedentary lifestyles and adverse nutritional habits. 85% to 90% of patients with hypertension have primary or essential hypertension (without an identifiable cause), while the remainder have secondary hypertension, of whom the majority have dysfunction of one or more endocrine systems [ ]. The most common causes of secondary hypertension are renal disease and endocrine disorders.
Although adrenal disease is often implicated as the cause of endocrine hypertension, pituitary as well as other extraadrenal disorders are also identified as the cause of the disease. Correct diagnosis of these disorders is of paramount importance because of the risk of potential complications in undiagnosed cases, the likelihood of optimal control with pharmacotherapy, and the probability of a surgical cure [ ]. Even though the prevalence of endocrine hypertension is relatively high compared to many other chronic illnesses, the diagnosis is often delayed or even never made because of inadequate awareness among physicians. This chapter attempts to provide an overview of some of the basic science aspects along with the clinical practice pearls about endocrine hypertension elaborated in other chapters of this textbook by the global experts in the field from twelve different countries across the five continents.

Primary hypertension versus secondary hypertension
Nearly 85% of patients with hypertension have primary (idiopathic or essential) hypertension, whereas the remaining 15% have secondary hypertension [ ]. The four common causes of secondary hypertension with their prevalence in the hypertensive subjects include obstructive sleep apnea (>5–15%), primary aldosteronism (PA) (1.4–10%), renal parenchymal disease (1.6–8.0%), and renovascular hypertension (1.0–8.0%) [ ]. Among patients with resistant hypertension, the prevalence of these four etiologies is >30.0%, 6.0–23.0%, 2.0–10.0%, and 2.5–20.0%, respectively. The less common causes include thyroid disorders, Cushing’s syndrome (CS), pheochromocytoma, coarctation of aorta, and drug-induced hypertension. A recently published retrospective analysis from China observed a change in the trend in secondary hypertension etiology [ ]. They observed that though renal parenchymal disease contributed to nearly 50% and obstructive sleep apnea to nearly 25% of hospitalized patients with secondary hypertension, there is a downward trend for renal parenchymal disease and an upward trend for obstructive sleep apnea, endocrine hypertension, renovascular hypertension, and aortic disease, from 2013 to 2016 ( P < .001).
The prevalence of secondary hypertension varies depending on age. The common causes in children are renal parenchymal disease and coarctation of aorta; in young adults are renovascular hypertension caused by fibromuscular dysplasia (especially in women), and renal parenchymal disease; in middle-age are PA, obstructive sleep apnea, CS, and pheochromocytoma; and in older adults are atherosclerotic renovascular hypertension, renal failure, and hypothyroidism [ ]. Marked discrepancies in the reported prevalence of secondary hypertension are due to the wide variations in the approach to diagnostic workup, inadequate awareness among physicians, and the resource strain for appropriate investigations in many population groups. For the same reasons, correct estimation of the actual prevalence of endocrine hypertension in the global population is difficult.
Once the clinical suspicion of endocrine hypertension arises, semiurgent diagnostic workup and the plan for appropriate management should be enforced to avoid catastrophic complications related to some of these disorders, with the probability of complete cure, or marked improvement of the disease, their complications, and hypertension. However, we must bear in mind that the clinical presentation of some of these disorders can be quite vague and can mimic many other systemic diseases, and therefore, the diagnosis is often delayed by several months to years. A thorough understanding of the basic science aspects, pathophysiology, clinical profile, diagnostic workup, and the management algorithm of endocrine hypertension are crucial for an appropriate scientific approach to patients with hypertension that we encounter in our day-to-day clinical practice. With the help of the most up-to-date evidence, the best global experts in the field of fundamental and clinical research elaborate on these aspects of endocrine hypertension in the subsequent chapters.
Individual chapters
Catecholamines and blood pressure regulation
Catecholamines (epinephrine, norepinephrine, and dopamine) are monoamine neurotransmitters and circulating hormones which play various vital roles in the maintenance of body homeostasis and BP regulation through different effector organs especially the cardiovascular system [ ]. These neurochemicals exert their wide variety of physiological and pathophysiological effects through three types of G-protein-coupled receptors viz. α-adrenoreceptors, β-adrenoreceptors, and dopamine receptors distributed throughout the body organs, tissues, and cells. While a steady physiological output of catecholamines is crucial for the maintenance of normal health, their excess production as seen in some patients with pheochromocytomas and paragangliomas (PPGLs), and deficiency states as in autonomic failure may be associated with catastrophic consequences. In their review of the role of catecholamines in BP regulation, Fernandez et al. provide a detailed analysis of these basic science aspects in Chapter 2 .
Adrenocortical hormones and BP regulation
Blood pressure in human beings is maintained in the normal physiological range through highly complex and interconnected mechanisms involving the symapthoadrenergic (central and peripheral) pathways that also include the chemoreceptors and baroreceptors, renin–angiotensin–aldosterone system (RAAS) that involves kidney and the adrenal cortical hormones (principally aldosterone), antidiuretic hormone from the hypothalamus, atrial natriuretic peptide (ANP) from the heart, and local factors from the blood vessel such as nitric oxide (NO) [ ]. Although the role of RAAS in BP regulation was known to man from the first half of the 20th century [ ], more light on this area was shed in our understanding during the latter half [ ]. Much more subsequent research input has been invested by the global scientific fraternity on to this important hormonal circuitry with many more newer discoveries that markedly changed our current approach to the management of hypertension.
Though aldosterone is the principal mineralocorticoid involved in the regulation of BP, other hormones of the adrenal cortex also play important roles in the human BP homeostasis. There is evidence to show the interrelationship between glucocorticoids [ ] and sex steroids [ ], and BP regulation in physiological and disease states. In their extensive review of the topic “Adrenocortical hormones and blood pressure regulation,” Sanders et al. provide us the current evidence on the mechanisms involved in this complex neuro-endocrine circuitry in health and disease ( Chapter 3 ). This chapter also highlights the importance of ongoing and future research in this area.
Hypothalamic–pituitary–adrenal axis and blood pressure regulation
Historically, the human hypothalamic–pituitary–adrenal (HPA) axis was an area of immense scientific research input in the past two centuries though we are still in an infantile stage of our understanding of its protean functions including those involved in human neurohumoral responses, endocrine homeostasis, and metabolic regulation. Ongoing research is expected to provide us with more rewarding output to unmask these mysteries in near future. The central regulation of BP is mainly through two unique neuro-endocrine circuitries, viz. the central sympathetic nervous system and the neuro-endocrine signals through the HPA axis while these two pathways are closely interlinked [ , , ]. In addition, the biological clock mechanisms in the brain and peripheral organs are also connected to these complex pathways and play significant roles in the neuro-endocrine signaling cascade which controls homeostatic equilibrium in healthy individuals. Various physiological, metabolic, hormonal, and psychological stressors can alter this equilibrium state that may affect BP regulation and thereby result in hypertension.
In their concise review “Hypothalamic-pituitary-adrenal axis and blood pressure regulation,” Pappachan, Fernandez, and Stratakis portray the complex mechanisms involved in the control of human BP in health and disease ( Chapter 4 ). Various factors that alter the physiological, psychological, and metabolic adaptive stress responses in health and disease states have been revealed in recent years [ , ]. However, considering the complexity and the current inadequate knowledge about these biological mechanisms, more research input is necessary for this area in the future as suggested by the authors of the chapter.
Renin-angiotensin–aldosterone system and blood pressure regulation
The history of RAAS began with the initial experiments of Tigerstedt and Bergman of the Karolinska Institute in 1898 showing the effects of rabbit’s renal tissue on arterial BP which led to the discovery of renin–angiotensin system in the early 20th century [ , ]. The puzzling effects of RAAS as an endocrine and neurohumoral effector circuit in the control of human health, well-being, and illness are still being evolved through the 21st century, improving some of our understanding of its complexities. Several physical, pharmaceutical, and surgical interventions can alter the functions of RAAS that may modulate homeostatic equilibrium and pathological states in man including hypertension, CVD, and infectious diseases like COVID-19 [ ].
The mechanisms of BP regulation by the RAAS are mostly elucidated in the recent years through decades of immense research. In their chapter “Renin–angiotensin–aldosterone system and blood pressure regulation,” Seravalle and Grassi outline the normal and pathophysiological mechanisms involved in RAAS and BP variations in health and disease ( Chapter 5 ). The role of RAAS in regulating systemic vascular resistance, and blood volume through the effects on salt-fluid balance, and vascular inflammation and remodeling is narrated in this chapter with up-to-date evidence. In addition, they highlight the importance of pharmacological modulation of RAAS as a key strategy in the treatment of hypertension, CVD, and chronic renal disease. The emerging role of mineralocorticoid antagonist therapy in the modern approach to management of resistant hypertension is also emphasized in this chapter.
Monogenic hypertension: an overview
The concept “monogenic hypertension” encompasses a group of genetic disorders associated with hypertension that follow a Mendelian inheritance pattern with a strong family history of the disease in affected individuals. Essential hypertension, on the other hand, is polygenic without a clear inheritance pattern. Although with some heterogeneity in its phenotypic expression depending on the degree of penetrance, monogenic hypertension usually presents with unique clinical and pathophysiological characteristics [ ]. With the help of modern DNA sequencing technology, several of the genes associated with monogenic hypertension are recently identified which improved our understanding of the pathobiology of these disorders making the clinical profiling, diagnostic evaluation, and management options easier. Most forms of monogenic hypertension syndromes are associated with dysfunction of sympathoadrenal or renal mechanisms of BP control owing to gain-of-function or loss-of-function mutations in the mineralocorticoid, glucocorticoid, or autonomic pathways [ ].
In their comprehensive and concise chapter “Monogenic Hypertension: An Overview,” Fernandez, Pappachan, and Scholl outline the classification, genetic associations, and diagnostic approach to monogenic hypertension ( Chapter 6 ). The algorithms demonstrated by the authors in this chapter along with a more detailed discussion on the genetic aspects of the inherited syndromes associated with hypertension in several other chapters of this textbook are expected to equip the readers with reasonable knowledge in working up and managing patients with endocrine hypertension. Ongoing research is expected to further improve our understanding of monogenic hypertension.
Primary aldosteronism (Conn’s syndrome)
PA results from overproduction of the mineralocorticoid hormone aldosterone from the adrenal cortex, usually from the zona glomerulosa layer. PA is the most common form of endocrine hypertension accounting for ≈10% of patients with elevated BP attending hypertension clinics and ≈20% of those with resistant hypertension [ , ]. Although the prevalence of PA is relatively high for a chronic disease state affecting a significant proportion of individuals in the society, the diagnosis of this condition in the community and even in the secondary care settings is alarmingly low because of the poor awareness of PA among the healthcare workers, difficulty with its diagnostic work-up in resource-poor settings in most underdeveloped economies, and the physician inertia to appropriately evaluate patients with hypertension to exclude PA. Correct identification and appropriate management of PA can potentially cure or improve the adverse outcomes associated with the disease, and therefore, a proactive approach from the endocrinologists to heighten the awareness of this enigmatic disease among healthcare providers is an urgent need of our time.
The chapter “Primary aldosteronism (Conn’s syndrome)” by Ceccato et al. brings forth the current evidence base on the clinical profile, diagnostic approach, and management algorithms with a brief discussion on the familial forms of PA to enable readers to have a better knowledge about the disease ( Chapter 7 ). Of the patients with PA, nearly one third only have unilateral disease with the remainder having bilateral adrenal involvement (with a small proportion having inherited monogenic forms of PA described below). A timely and accurate diagnosis of PA is of paramount importance as resection of an aldosterone-producing adenoma (APA) is associated with a cure of hypertension in 50 to 60% of individuals, and with significant improvement in the remainder [ , ]. However, the correct identification of cases through appropriate biochemical workup and subtyping the disease through investigations such as adrenal venous sampling (AVS) in those with PA often pose significant challenges to clinicians. Through the detailed and precise narrative of the diagnostic and therapeutic approaches to PA in their chapter, Ceccato et al. enable the readers to overcome these challenges.
Familial hyperaldosteronism
The genetic landscape of PA has been largely disclosed in recent years making use of next-generation sequencing (NGS) methods which identify several somatic and germline mutations improving our understanding of the pathobiology of the disease [ ]. Familial hyperaldosteronism (FH) forms a group of monogenic hypertensive disorders associated with PA that shows a clear inheritance pattern with phenotypic variability depending on the degree of penetrance of the culprit gene. There are 4 main forms of FH (FH-I, II, III, and IV) currently known, with an additional disorder known as PASNA (PA with seizures and neurological abnormalities) syndrome described recently. Because of the dominant inheritance nature, patients with FH present early in life (usually before the age of 20 years) with hypertension and sometimes a hypertensive complication such as hemorrhagic stroke. In their chapter “Familial Hyperaldosteronism,” Pappachan, Fernandez, and Geller discuss the pathobiological aspects, diagnosis, and management of FH with the aid of latest research evidence ( Chapter 8 ).
Congenital adrenal hyperplasia and hypertension
Congenital adrenal hyperplasia (CAH) is a group of genetic disorders characterized by abnormalities in the enzymes catalyzing adrenal steroidogenesis. CAH is inherited as an autosomal recessive disorder, and there are 7 different types depending on the enzyme deficiency involved, viz. 21-hydroxylase (21OH), 11β-hydroxylase (11βOH), 17α-hydroxylase/17,20-lyase (17OH), 3β-hydroxysteroid dehydrogenase type 2 (3βHSD2), steroidogenic acute regulatory protein (StAR), P450 cholesterol side-chain cleavage enzyme (P450scc), and P450 oxidoreductase (POR) [ ]. CAH secondary to 21-OH deficiency (from CYP21A2 mutation) is the most common form of the disease accounting for ∼95% of cases with an incidence of ∼1 in 15,000, and is not usually associated with hypertension as a part of the primary disease [ ]. Of the CAH subtypes, those due to 11β-hydroxylase and 17α-hydroxylase deficiencies are associated with hypertension in the affected individuals [ ]. Very rarely POR deficiency may also result in hypertension in the affected individuals [ ]. However, among patients with monogenic hypertension, CAH accounts for only a small proportion of cases.
In their chapter “Congenital Adrenal Hyperplasia and Hypertension,” Gurpinar and Guran provide a detailed account of these uncommon disorders of monogenic hypertension that often presents during childhood ( Chapter 9 ). However, we must bear in mind that hypertensive forms of CAH can also be associated with variable phenotypic expression, and therefore, some of these patients may present to the clinician for the first time with hypertension during early adult life. In their detailed monograph, the authors provide us with the clinical features, diagnostic approach, management algorithms, and follow-up plan for patients with these subtypes of CAH associated with hypertension.
Endocrine hypertension: discovering the inherited causes
As mentioned in the section on monogenic hypertension, there are several inherited monogenic disorders which result in endocrine hypertension. More than 30 genes and ∼1500 single nucleotide polymorphisms have been identified to date in association with monogenic forms of hypertension or hypotension syndromes which mainly involve abnormalities in the RAAS and adrenal steroidogenic pathways and some uncommon neuroendocrine tumors [ ]. Each of these endocrine hypertensive disorders may invoke enthusiasm in the readers to understand their pathophysiology, clinical profile, diagnostic evaluation, and management algorithm. Assadi and colleagues in their chapter “Endocrine hypertension: discovering the inherited causes” do that job in a very concise and precise way ( Chapter 10 ) summarizing the complex topic with the aid of multiple tables and figures.
Pheochromocytomas and hypertension
Although the prevalence of PPGLs among patients with adrenal incidentalomas is 5–7% [ ], they are rare endocrine tumors with an estimated prevalence of 2 to 8 per million in the general population [ ] and account for only 0.1–0.6% of cases of hypertension attending general medical outpatient clinics [ , ]. PPGLs are unique in their secretory behavior with vasoactive catecholamine production and often present with sustained and/or episodic hypertension. The molecular and genetic landscapes of PPGLs are clearer in recent years with the discovery of several mutations and tumor syndromes which give us a better understanding of their clinical, epidemiological, and pathobiological characteristics to enable the development of more scientific diagnostic, prognostic, and management algorithms [ ]. With the aid of up-to-date evidence, Goemann and Maia elaborate on these algorithms in their comprehensive chapter “Pheochromocytomas and hypertension” ( Chapter 11 ).
Paragangliomas and hypertension
Paragangliomas (PGLs) are rare neuroendocrine tumors originating from the embryonic neural crest cells which form the extra-adrenal autonomic paraganglia dispersed from the skull base to the pelvic floor [ ]. Though many of the clinical, pathophysiological, and genetic nature simulate those of adrenal pheochromocytomas, PGLs show certain unique clinical, familial, and pathophysiological characteristics [ ]. These include sustained arterial hypertension or rarely normotension (dopamine-secreting tumors), malignant/metastatic potential, genetic characteristics (predominantly Cluster 1-related gene mutations), and relatively silent clinical behavior (often detected as incidentalomas in imaging studies or during open surgery). Majority of the functional PGLs (70–80%) arise from the infradiaphragmatic sympathetic ganglia, whereas the head and neck PGLs are rarely hormone producing. With the help of a couple of case vignettes, Petrák and Zelinka give us a detailed account of PGLs in their chapter “Paragangliomas and hypertension” enabling the readers to have a very thorough understanding of these rare neoplasms causing endocrine hypertension ( Chapter 12 ).
ACTH-dependent Cushing syndrome
CS resulting from glucocorticoid administration for pharmacotherapy of various disease states is common. However, endogenous CS is a rare endocrine disorder associated with protean manifestations that may vary from subclinical hypercortisolemia without significant major clinical abnormalities to florid multiorgan dysfunction and cushingoid appearance. Approximately 70–80% of endogenous CS cases result from excess production of adrenocorticotrophin (ACTH) from the pituitary (≈60–70%; Cushing’s disease/CD) or from an extrapituitary source (5–10%), while the remainder are caused by primary adrenal disorders [ ]. Endogenous hypercortisolemia resulting from ACTH overproduction is termed ACTH-dependent CS. Hypertension is the most common sequel of CS regardless of the etiology and can occur in 75–85% of adults and 47–51% of children and adolescents [ , ]. The severity of hypertension in patients with CS may be related to multiple factors including the age of the patient and the duration and degree of cortisol exposure [ ]. However, the prevalence of CS in hypertensive population is relatively low at 0.5%, and only <1% of cases with resistant hypertension result from CS [ ].
The pathophysiological factors involved in the causation of hypertension in patients with CS are not fully elusive though various mechanisms are proposed to explain this important comorbidity, mostly based on small-scale studies [ ]. The diagnosis and management of CS can often be challenging and require close multidisciplinary association with different specialties such as biochemistry, radiology, surgery, and endocrinology. A recently updated international consensus on evaluation and management of CD gives us insight into the complexities of this approach [ ]. Currently, there are no guidelines exclusively for the management of hypertension in patients with CS, and therefore, we must rely upon the general guidelines for managing essential hypertension in any patient to treat such cases. In their concise and comprehensive chapter “ACTH-dependent Cushing’s syndrome,” Fernandes, Varlamov, and Fleseriu successfully fill these evidence gaps ( Chapter 13 ).
Adrenal Cushing’s syndrome
Approximately, 20–30% of endogenous CS is ACTH-independent resulting from adrenal disease (adrenal CS) as mentioned above. The annual incidence of adrenal CS is 1.3 per million as reported by a large recent series from Korea [ ]. Adrenal CS results from cortisol producing adrenal adenoma (most common; >50%), adrenocortical carcinoma, and bilateral micro and macronodular adrenal hyperplasia [ ]. Several genetic disorders and syndromes have been implicated in the causation of these types of CS [ ]. In his chapter “Adrenal Cushing’s syndrome”, Ragnarsson provides us with the latest evidence on epidemiology, genetic aspects, clinical profile, diagnostic evaluation, pathophysiology of hypertension, and management of patients with adrenal CS ( Chapter 14 ).
Hypertension in growth hormone excess (acromegaly) and deficiency
Growth hormone excess that results in acromegaly had been a puzzling hormonal disorder for modern medical practice historically, and continues to pose significant diagnostic and management challenges even in the 21st century. Acromegaly is a rare endocrinopathy with a disease prevalence of 28–137 cases per million and an annual incidence ranging between 2 and 11 cases per million people [ ]. Although hypertension is the most common comorbidity among patients with acromegaly, this endocrine disorder contributes only a negligible proportion to the global hypertensive population. Recent estimates show a hypertension prevalence of 11–55% in patients with acromegaly [ ]. Hypertension in acromegaly is multifactorial and the mechanisms involved are increased plasma volume from fluid retention, increased vascular resistance, and endothelial dysfunction [ ], along with other factors such as sleep apnea, insulin resistance, and increased cardiac output [ ]. Hypertension is one of the most important comorbidities in acromegaly associated with excess mortality from associated CVD.
Adult growth hormone deficiency (AGHD) on the other hand is also associated with a high risk of CVD mainly because of the development of metabolic syndrome, an integral component of which is hypertension [ ]. AGHD is a rare disease with an estimated annual incidence of 16.5 per million population [ ]. Therefore, AGHD also accounts for a negligible proportion of patients with hypertension in the general population. As in acromegaly, the pathophysiology of hypertension in AGHD also is not fully understood and various mechanisms have been proposed by different authors.
In their detailed review of the currently available literature for diagnosis and management of acromegaly and growth hormone deficiency (GHD), Mihai and Korbonits provide extensive scientific evidence-base to enable clinicians to approach these uncommon endocrine problems. Therefore, the chapter “Hypertension in patients with growth hormone excess and deficiency” ( Chapter 15 ) is an invaluable resource to this textbook.
Hypertension in thyroid disease and hyperparathyroidism
Thyroid disease forms the second most common type of endocrine disorder after diabetes mellitus, while primary hyperparathyroidism forms the third. Large studies show the prevalence of hypothyroidism at 4–7% [ ], hyperthyroidism at 0.5–2.1% (overt hyperthyroidism 0.1–0.5%) [ ], and primary hyperparathyroidism at ≈1% of adults [ ] among the general population. Both types of thyroid diseases and primary hyperparathyroidism are directly associated with an increased prevalence of hypertension. Considering the relatively high prevalence of these endocrinopathies in the population, they constitute important causes of hypertension, and therefore, a thorough understanding of the pathophysiology, clinical presentation, and management of thyroid disease and primary hyperparathyroidism is crucial while approaching patients with endocrine hypertension. Szwarcbard and Topliss in their chapter “Hypertension in thyroid disease and hyperparathyroidism” provide readers with this wonderful opportunity ( Chapter 16 ).
Obesity, insulin resistance, and obstructive sleep apnea
The prevalence of obesity in the global population has been steadily increasing over the past 5–6 decades because of the adverse lifestyles and modernization of food industry that resulted in a fast-food culture across the world. Numerous complex mechanisms have been proposed to explain how overweight and obesity result in hypertension [ ]. One of the key components of metabolic syndrome, commonly observed in obese individuals, is hypertension. There is also clear evidence to show that weight gain usually increases BP, while weight loss is associated with reduction of BP [ ]. Obesity-related primary hypertension was found to affect 78% and 65% of men and women, respectively, in the Framingham Heart Study [ ]. Obesity also results in insulin resistance and obstructive sleep apnea (OSA), the other important risk factors for the development of hypertension.
OSA, the commonest form of sleep-disordered breathing, increases the risk of hypertension by several mechanisms. The estimated prevalence of OSA is 15–24% among the adult population [ ]. Considering the inadequate awareness among healthcare professionals about the high prevalence of the disease, and the inertia for appropriate diagnostic evaluation from physicians, there is a high likelihood of under-reporting of OSA. In addition to the higher risk of essential hypertension related to obesity, patients can also develop OSA-related secondary hypertension (ORSH), often resistant to usual pharmacotherapy [ ]. Clear understanding of the pathophysiology, clinical presentation, diagnostic evaluation, and management of OSA and ORSH is important in managing patients with endocrine and secondary hypertension. This task is very efficiently performed by Oduro-Donkor and Barber in their chapter “Obesity, Insulin Resistance and Obstructive Sleep Apnea” to enable the readers of this textbook to look for and to treat OSA, if present, in individuals with resistant hypertension ( Chapter 17 ).
Endocrine hypertension in children
Hypertension is becoming a major health problem among children and adolescents with a recently estimated prevalence of 4% in those younger than 19 years [ ]. Approximately 60% of cases of hypertension in children are from secondary causes such as CVD and renal disorders [ ]. Endocrinopathies as a cause account for only 0.05–6% of cases of hypertension in the pediatric population [ ]. Therefore, a more vigilant clinical and diagnostic approach should be practiced by the clinicians while dealing with these patients as the signs and symptoms can be subtle, and may not be often familiar to many professionals. Prompt suspicion and urgent clinical workup of all children with hypertension are mandatory to avoid hypertension-related complications including premature mortality.
The pathophysiological features, clinical characteristics, and laboratory parameters of endocrine hypertension in childhood and the approach for diagnostic workup are also often different from those in the adult population with the disease. Reading the chapter “Endocrine Hypertension in Children” by Valaiyapathi and Ashraf ( Chapter 18 ) would enable us to obtain a clear evidence-based understanding of how to approach and manage children and adolescents with these uncommon disorders associated with high morbidity and mortality.
Endocrine hypertension in pregnant woman
Pregnancy is a physiological state with marked variations in the maternal body metabolism, circulation, and chemical composition to accommodate the physiological needs of the growing fetus. Therefore, there shall be significant variations in the neurohumoral and hormonal balance in a pregnant woman compared to a nonpregnant female. Output of normal female hormones like estrogens and progesterone shall be very different during pregnancy compared to nonpregnant states, and several placental hormones such as human placental lactogen and human chorionic gonadotropin also come into play in the maternal circulation [ ]. Marked alterations in the HPA, and hypothalamic–pituitary–gonadal (HPG) axes, also occur during pregnancy compounding the situation further [ ]. Moreover, there shall be mechanical factors such as compression of other abdominal organs including the kidneys and the adrenals by the gravid uterus that may result in exacerbation of the preexisting diseases of these organs present prior to pregnancy.
Approximately, 30% increase in the blood and plasma volume occur during pregnancy, and therefore, the hormone assays are expected to have different cut-offs especially with the interference from alterations in the HPA/HPG axes and placental hormones as mentioned above. Several of the imaging modalities utilizing ionizing radiations (e.g., CT scan) and compounds (e.g., nuclear imaging techniques) are contraindicated during pregnancy, and some of the pharmacotherapeutic agents used for the management of hypertension are unsafe. All these issues make diagnosis and management of endocrine hypertension during pregnancy complicated in ordinary clinical settings. To address these issues, and to empower readers with the best evidence, Jebasingh and Thomas compiled their chapter “Endocrine Hypertension in Pregnancy” which has turned out to be an invaluable resource ( Chapter 19 ) to this textbook.
Imaging for patients with endocrine hypertension
Imaging studies for confirmation of the disease are integral and final components of the diagnostic algorithms in hormone science. Full laboratory workup to reach the biochemical diagnosis is almost always followed by imaging studies to identify the structural and/or functional abnormalities in the endocrine glands which help us to plan definitive treatment that may lead to a surgical cure of the disease. Endocrine hypertension is often caused by tumoral involvement of one or more of the hormone-producing glands which can be detected by an anatomical imaging modality such as CT, MRI, or ultrasonography scans for disease localization. For example, primary hyperaldosteronism is caused by an APA in about one third of individuals which may be localized by a CT scan of the adrenal gland with subsequent lateralization (for aldosterone overproduction) study by an image-assisted AVS to pinpoint the site for surgical target [ ]. Adrenalectomy in such cases is associated with a cure rate of hypertension in 50–60% and improvement in most of the remainder [ ].
Another important group of imaging studies which assist final diagnosis in patients with endocrine hypertension is functional imaging to localize the abnormal gland(s) causing hormone overproduction. Several nuclear imaging techniques utilizing radioisotopes have been made use of in the past few decades for imaging the adrenal, thyroid, parathyroid, and pituitary glands for disease localization/detecting functional hyperactivity. For example, 68 Ga-DOTATATE PET/CT scan has emerged as the best imaging modality in recent years for the detection and staging of neuroendocrine tumors with a reported sensitivity of 93% and specificity of 95% [ ]. Anatomical and functional imaging studies are also important for prognostication and follow-up management of patients after initial treatment. For example, 123 I-MIBG ( 123 Iodine-labeled metaiodobenzylguanidine)-avid malignant pheochromocytomas are more likely to respond to radionuclide therapy with 131 I-MIBG [ ]. Basic understanding of the relevant imaging studies and adequate knowledge about diagnostic performance of each imaging modality are essential among physicians who workup patients with endocrine hypertension for appropriate patient care and optimal resource utilization. With the back-up of up-to-date evidence through extensive literature review and several good quality images, Ordidge and Sahdev did a wonderful job through their chapter “Imaging for Patients with Endocrine Hypertension” ( Chapter 20 ) in enabling the readers of the textbook to gain thorough ideas on “whom,” “what,” and “when” to image when endocrine hypertension is suspected.
Systematic approach for the diagnosis and management of endocrine hypertension
“Last but not the least chapter” of this textbook is “Systematic Approach for the Diagnosis and Management of Endocrine Hypertension” by Stowasser, Jansen, and Wolley ( Chapter 21 ). A structured and systematic approach to any clinical problem is expected to give the best answer to every puzzling disorder in our day-to-day medical practice in the diagnostic workup and management options. This approach is very crucial in practicing hormone science with a special emphasis on working up and treating patients with endocrine hypertension considering the diagnostic and therapeutic complexities involved in such cases. For example, diagnosis and disease localization in PA can be often difficult and a vast majority of cases in the community, and even in the secondary care settings, are underdiagnosed considering the relatively high prevalence rates of ≈10% in patients attending hypertension clinics and ≈20% with resistant hypertension [ ]. Moreover, there is ongoing debate even among endocrinologists about the screening criteria for patients, the appropriateness of individual cut offs regarding plasma renin activity (PRA), direct renin concentration (DRC), plasma aldosterone concentration (PAC), and aldosterone renin ratio (ARR), the need for AVS in each case for definitive therapeutic planning and individualizing medical therapy [ ].
To add more complexity to the above conundrum regarding the approach to patients with PA, several authors also reported marked ethnic variations in the RAAS activity with differences in the plasma levels of renin and aldosterone especially in African and African American ethnic groups [ ]. This fact would demand ethic-specific approach to patients and the necessity of changing the current international guidelines accordingly. With their extensive experience in developing real-world practical algorithms for the diagnosis and management of PA in Australia, backed up by the latest and extensive literature review, Stowasser et al. fill in the evidence gap in this controversial area at large through Chapter 21 .
An individualized approach to patients with hormonal disorders is highly important for planning diagnostic workup and management while dealing with cases of endocrine hypertension. In the subsequent sections of the chapter, this task has been very effectively performed by Stowasser et al. For example, by the discussion regarding the change in age-standardized incidence rate for the incidence of PPGLs, the authors provide guidance to enable an individualized approach while reviewing patients [ , ]. Overall, Chapter 21 would serve as a quick reference guide within this textbook to empower the readers with the best evidence to manage their patients with endocrine hypertension.
Recent trends/emerging concepts
Many of the conditions associated with endocrine hypertension have a genetic/familial background of inheritance that makes it essential for appropriate testing to prognosticate/follow-up the patients and families with the disease gene. These tests can be labor-intensive and expensive, and therefore, endocrinologists should use them very judiciously. Recent guidelines and emerging testing algorithms help us to approach such cases more pragmatically as discussed briefly in the following sections.
Genetic testing for familial PPGL
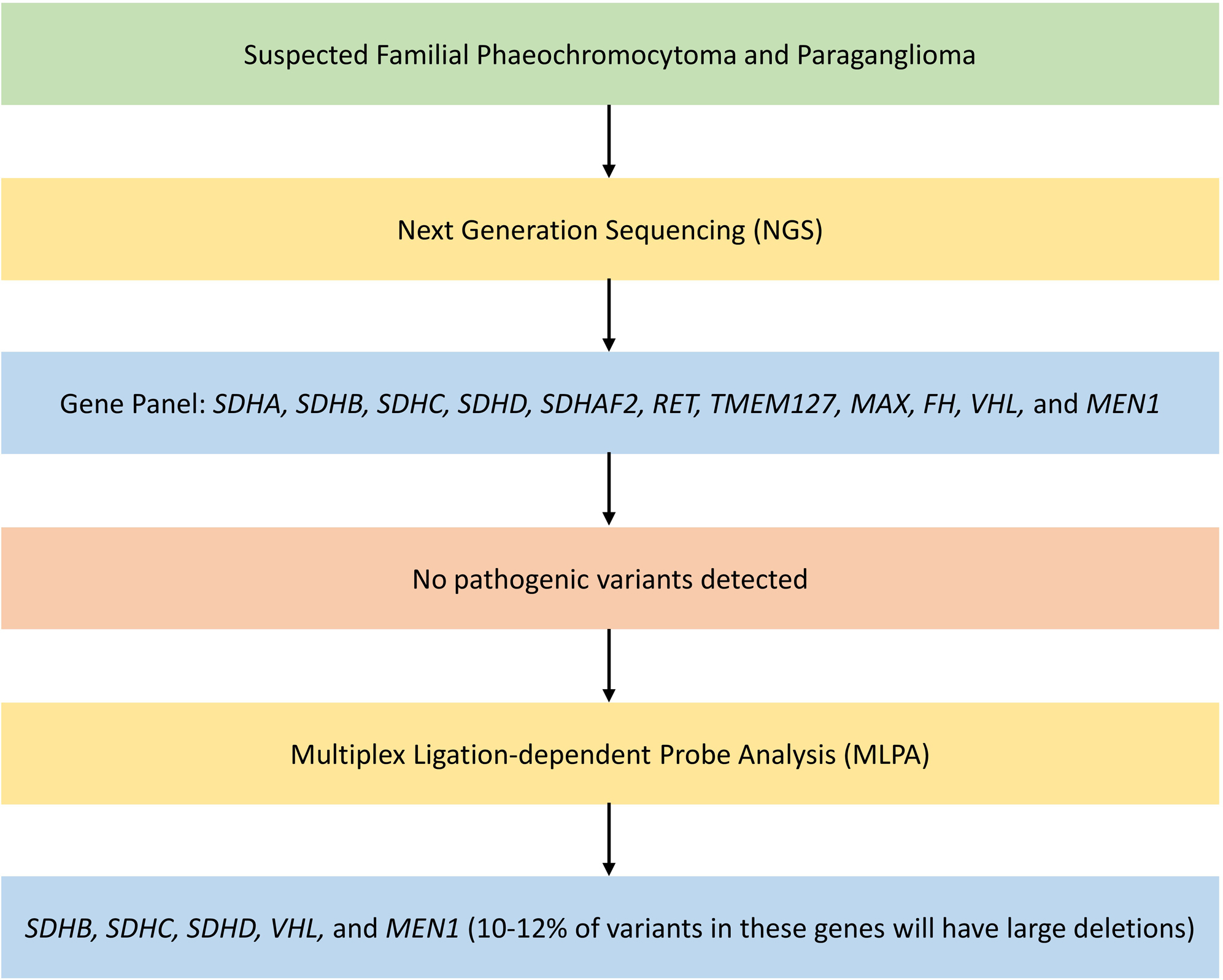
Large number of susceptibility genes are implicated in the diagnosis of familial PPGLs. NGS technology is ideally suited for genetic testing of patients with PPGL [ ]. NGS involves DNA sequencing using a disease-specific gene panel including SDHA, SDHB, SDHC, SDHD, SDHAF2, RET, TMEM127, MAX, FH, VHL, and MEN1 [ ]. The gene panel used might vary between laboratories: basic panel—looking for germline mutations with highest evidence, extended panel—looking for germline mutations at basic panel genes and at candidate susceptibility genes with low evidence, and comprehensive panel—looking for somatic mutations in addition to the extended panel [ ]. Any sequence variants observed would be classified as pathogenic, benign, or variants of uncertain significance (VUS) by the reporting laboratory. NGS has high risk of false positivity as they detect many VUS [ ].
NGS is not capable of detecting large deletions or insertions, thereby causing false negative results [ ]. Multiplex ligation-dependent probe analysis (MLPA) together with NGS can improve the sensitivity [ , ]. If no pathogenic variants are detected on NGS, then MLPA should be done for SDHB, SDHC, SDHD, VHL, and MEN1 as 10%–12% variants in these genes will have large deletions [ ]. Germline mutations in SDHA, SDHB, SDHC, and SDHD account for 30% of all hereditary PPGL cases [ ]. They are associated with high risk of malignancy, with increased lifetime risk of multifocal and synchronous tumors. These mutations predispose to PPGL, gastrointestinal stromal tumor (GIST), renal cell carcinoma (RCC), and pituitary tumors.
Indications for genetic testing in PPGL as per NHS England national genomic test registry are as follows (if patient meets one of the criteria): [ ]
- 1.
All patients <60 years with a diagnosis of pheochromocytoma
- 2.
Patients at any age with bilateral pheochromocytoma
- 3.
Patients at any age with pheochromocytoma and RCC
- 4.
Patients at any age with any paraganglioma
- 5.
Patients with pheochromocytoma/paraganglioma with loss of staining for SDH proteins on immunohistochemistry
- 6.
Patients at any age with pheochromocytoma/paraganglioma and a family history of pheochromocytoma/paraganglioma/RCC/GIST
Fig. 1.1 shows an algorithm for genetic testing for familial PPGL.