Abstract
The reproductive axis, particularly in women, is vulnerable to disruption from environmental influences and illness. Endocrine disorders not involving the gonads strongly influence reproductive function. Diseases of the pituitary, adrenal, and thyroid can exert deleterious effects on ovulation, sex steroid production, implantation, and pregnancy outcomes. This chapter will review the presenting features, evaluation, and treatment of major endocrine disorders that affect reproduction, with emphasis on mechanisms that impair fertility.
Keywords
Pituitary, hyperprolactinemia, adrenal, Cushing syndrome, Cushing disease, hypothyroidism, thyroid, hyperthyroidism, congenital adrenal hyperplasia
The reproductive systems are vulnerable to disruption by internal and external forces including disease, malnutrition, and various forms of stress. The male and female axes are both susceptible to dysfunction from the same processes, although the female axis tends to be more sensitive. This chapter will review the influence of endocrine disorders of the pituitary, adrenal, and thyroid on reproduction. Each section will discuss the most important and relevant disorders of the specific gland and its effects on female reproduction first. Features specific for male reproduction will be discussed where relevant in each disease subsection. The reader is directed to Chapter 3 , Chapter 17 , Chapter 20 for review of neuroendocrine disorders; Chapter 4 , Chapter 16 , Chapter 17 for further discussion of congenital adrenal hyperplasia (CAH); Chapter 21 for a review of hyperandrogenism; and Chapter 27 for a review of endocrine disorders in pregnancy.
Pituitary Disorders
- ◆
Hypopituitarism from pituitary tumors and trauma tends to follow an order of hormone loss: first growth hormone (GH), then luteinizing hormone (LH)/follicle-stimulating hormone (FSH), followed by thyroid-stimulating hormone (TSH), and finally adrenocorticotropic hormone (ACTH).
- ◆
Serum prolactin elevations from stalk compression rarely exceed 250 ng/mL.
- ◆
Indications for dopamine agonist therapy of microprolactinomas include galactorrhea, infertility, and estrogen deficiency; oral contraceptive pills are an alternative only for estrogen deficiency.
Overview
As the “master gland,” the anterior pituitary gland controls the secretion of several essential hormones from other major endocrine glands, including the thyroid (releasing thyroxine and triiodothyronine), adrenal cortex (cortisol, dehydroepiandrosterone sulfate [DHEAS]), and gonads (predominantly estradiol plus progesterone in females and testosterone in males). The anterior pituitary gland also produces GH and prolactin, which act directly on target organs. The actions of GH are largely exerted via the local or systemic production of insulin-like growth factor-1 (IGF1). The posterior pituitary regulates water metabolism via the production of vasopressin and induces milk letdown via the production of oxytocin. Disorders of the pituitary can be partial or complete, isolated to one hormone or multiple, and related to hormone deficiency or hormone excess. The axes controlled by the pituitary gland share the following basic principles:
- 1.
Input from higher brain centers to the hypothalamus
- 2.
Releasing and inhibitory factors influencing pituitary hormone secretion
- 3.
Hypothalamic factor pulsatility, leading to pulsing of pituitary and target gland hormones
- 4.
Feedback inhibition at both the hypothalamus and pituitary by active target gland hormones
- 5.
Peripheral metabolism of target gland products
- 6.
Influence of diurnal rhythm
The relative importance of these regulatory components varies for each axis and is described below in more detail. These axes are shown schematically in Fig. 24.1 .
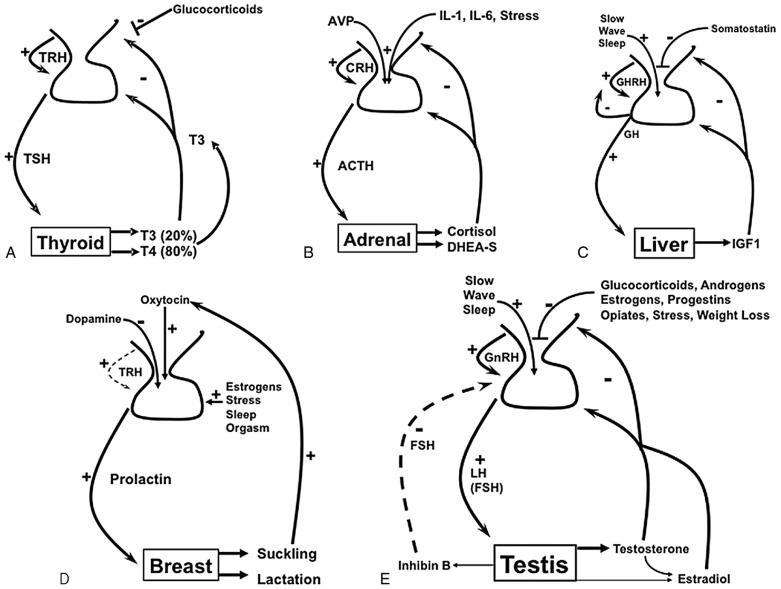
Of the pituitary axes, the thyroid axis is the simplest for several reasons. The primary product of the thyroid is thyroxine (T4), which is a precursor of the active hormone triiodothyronine (T3). Since T4 is heavily protein-bound, T4 has a long half-life (7 days), and T4 is slowly metabolized to T3. This scenario provides steady, well-dampened feedback, and the thyroid axis therefore shows high stability and low pulsatility. Hypothalamic thyrotropin-releasing hormone (TRH) pulses stimulate release of TSH; however, negative feedback by T3, primarily the pool generated via de-iodination of circulating T4 in the pituitary, tightly regulates TSH biosynthesis and primarily regulates the thyroid axis. Thus the thyroid axis is a model endocrine feedback system due to its stability and simplicity.
In contrast, the adrenal axis is characterized by a strong diurnal rhythm, with the shorter half-life of cortisol (1 hour) leading to greater pulsatility. The adrenal axis is also more sensitive to factors beyond hypothalamic corticotropin-releasing hormone (CRH) and negative feedback by cortisol. Both vasopressin and various cytokines increase production of corticotropin (ACTH) in response to stress or illness. As a consequence, this intricate feedback system provides a more responsive axis to physiologic changes and needs. This level of sophistication, however, complicates clinical testing.
The GH (somatotropin) axis is primarily a two-component axis regulated by the hypothalamus. Growth hormone–releasing hormone (GHRH) serves as the major positive stimulus and somatostatin (SS) as the major negative stimulus. GH stimulates the production of IGF1 in the liver and locally in other tissues. Circulating IGF1 derives from the liver and exerts some negative feedback on the axis, but this influence is small relative to SS. Numerous hormonal and metabolic factors, however, also alter GH secretion by modulating the release of GHRH and SS. While the two hypothalamic hormones regulate GH pulsatility throughout the day, the greatest GH pulses are produced in slow-wave sleep. Significant physical stressors, including hypoglycemia, also increase GH secretion.
Prolactin is the only anterior pituitary hormone that is primarily under negative control, mediated by dopamine. For this reason, prolactin rises whenever blood flow to the pituitary from the hypothalamus is impaired. Therefore, hyperprolactinemia can result from a large pituitary tumor, which does not itself secrete prolactin, by blocking blood flow from the stalk. Prolactin is also increased by stress, nipple stimulation, and TRH, so prolactin rises transiently for several reasons and persistently for several others ( Table 24.1 ). Prolactin acts on the breast to enable lactation, but there is no known hormonal product from the breast that exerts negative feedback on prolactin secretion. Estrogens also stimulate lactotrope growth.
| Cause | Characteristic Features(s) |
|---|---|
| Prolactinoma | Mass effects if macroadenoma |
| Acromegaly | Headaches, heavy perspiration, acral changes |
| Macroadenoma (not prolactin secreting) | Peripheral vision loss, anterior pituitary defects |
| Other infiltrative or hypothalamic diseases | Anterior pituitary defects, can have diabetes insipidus |
| Drugs | Other specific side effects of drug |
| Pregnancy | Positive human chorionic gonadotropin, amenorrhea |
| Renal failure | Comorbidities of renal failure |
| Chest wall stimulation | Variable prolactin |
| Stress | Including phlebotomy |
| Primary hypothyroidism | See Table 24.4 |
The gonadotropins LH and FSH and their regulation by gonadotropin-releasing hormone (GnRH) are discussed in detail for the male and female in other chapters. Here we simply emphasize that pulsatile GnRH secretion every 90 to 120 minutes is critical to LH and FSH production. While this principle is true for both males and females, the influence on fertility and symptoms differs in other respects. The reproductive axis is particularly sensitive to disorders and disruptions of the pituitary-hypothalamic axes as discussed in further detail later.
Loss of pituitary function is most commonly caused by drugs, exogenous hormones, or tumors. Tumors affect pituitary function by overproduction of hormones, such as prolactin impairing gonadotropin secretion and hypercortisolism lowering TSH and gonadotropins, or by mass effect. Any pituitary tumor greater than 1 cm in maximal diameter is defined as a macroadenoma. The acquisition of pituitary hormone deficiencies due to tumors tends to follow the order GH first, then LH + FSH, then TSH, and finally ACTH. Consequently, the reproductive axis is fairly vulnerable to disruption by macroadenomas of any cell type. Decompression by transsphenoidal surgery can restore pituitary function, particularly for the ACTH and TSH axes, but iatrogenic hypopituitarism is a common risk of surgery. Radiotherapy tends to cause hypopituitarism over a period of 2 to 15 years and for this reason should be used judiciously in women of reproductive age. In the following discussion, both the nature of the pituitary disorder and its treatment are addressed as they affect strategies for restoring reproductive function.
Pituitary Disorders That Affect Reproduction
Prolactinoma and Hyperprolactinemia
The combination of amenorrhea and galactorrhea in a young woman is a classic presentation of prolactinoma; however, these two symptoms may occur individually or not at all. Hyperprolactinemia has many potential etiologies (see Table 24.1 ) (see Chapter 3 ). The mechanisms of reproductive dysfunction in hyperprolactinemia vary somewhat with etiology, but prolactin disrupts the pulses of GnRH and also directly reduces the production of LH and FSH.
The diagnosis of hyperprolactinemia is established by measuring a serum prolactin at any time of day without dynamic testing, although the stress of phlebotomy can cause slight elevations. Falsely elevated prolactin measurements can be caused by the presence of macroprolactin, also known as big-big prolactin, which is a complex of prolactin and IgG detected variably in different immunoassays but lacking normal biological activity. If the prolactin is clearly elevated and causes other than pituitary tumors are excluded, a dedicated magnetic resonance image (MRI) of the sella with gadolinium contrast agent should be performed.
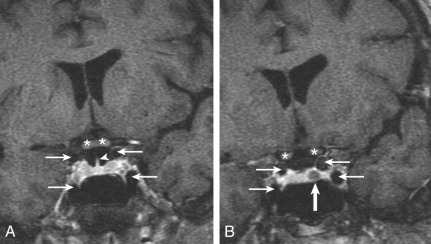
Microprolactinomas (tumors <1 cm in greatest diameter) are found in about 1% of women ages 20 to 40 years old. The degree of prolactin elevation is roughly proportional to the size of the tumor. A tumor not secreting prolactin might cause hyperprolactinemia via stalk compression and impaired dopamine delivery, but the prolactin rarely if ever rises above 250 ng/mL. For example, a patient with a 3-cm pituitary mass and a prolactin of 150 ng/mL probably does not have a prolactinoma. In cases of large pituitary tumors with mild prolactin elevations, however, the prolactin measurement should be repeated with dilutions to identify the “high-dose hook effect,” which artifactually lowers the assayed value. GH is a full prolactogen in humans; consequently, galactorrhea with mildly elevated prolactin and a pituitary tumor could be secondary to a somatotropinoma rather than prolactinoma. On T1-weighted MRI with gadolinium contrast, microprolactinomas tend to be hypointense (hypoenhancing) relative to the normally bright pituitary and usually do not distort the architecture of the gland ( Fig. 24.2 ). Although the larger macroprolactinomas tend to enhance with gadolinium, the pattern is quite variable, and macroprolactinomas tend to distort the architecture of the gland ( Fig. 24.3 ) with the inferior portion of the pituitary stalk often deviating away from the tumor.
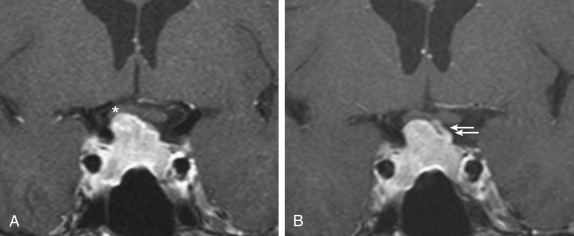
Prolactinomas are generally very responsive to medical therapy with dopamine agonists (bromocriptine and cabergoline). Even for large and invasive macroprolactinomas with visual changes, tumor shrinkage by dopamine agonists can be immediate and effective (see Fig. 24.3 ). Treatment options are selected based on symptoms, tumor size, and patient goals. Indications for treatment include infertility, amenorrhea, galactorrhea (particularly if spontaneous and bothersome), hypopituitarism, and mass effect. A woman with a 5-mm microprolactinoma, serum prolactin of 60 ng/mL, regular menses, and only trace expressible galactorrhea does not require treatment, because these tumors rarely grow. In contrast, the same woman with infertility would be treated if she desires pregnancy. Cyclic estrogen and progestin for endometrial and bone protection or symptoms of estrogen deficiency is an option for patients with microprolactinomas and irregular menses but without galactorrhea and who do not desire pregnancy, with low risk of tumor growth.
Bromocriptine is administered in two to three divided doses with a total of 2.5 to 40 mg/day. The main side effects of bromocriptine are nausea, lightheadedness, and nasal stuffiness. The dose is slowly advanced every 4 to 10 days as tolerated until the serum prolactin is normalized and the symptoms are relieved. Cabergoline, administered at 0.25 to 2 mg once or twice weekly, is much more potent and better tolerated than bromocriptine. Bromocriptine is generally preferred if pregnancy is desired, stopping the medication upon confirming conception. Cabergoline use early in pregnancy was not found to increase the risk for miscarriage or fetal malformations and is generally considered safe also. Visual fields and serum prolactin should be monitored throughout pregnancy and during lactation, more closely for macroprolactinomas. MRI without contrast is performed if tumor growth is suspected, and cabergoline is restarted to prevent vision compromise and/or hypopituitarism during pregnancy.
In some cases, particularly those in which the tumor has become no longer visible on MRI scan, cabergoline can be discontinued after 2 to 5 years without recurrence of hyperprolactinemia. Chronic treatment with high doses of cabergoline for Parkinson disease has been rarely associated with cardiac valve disease due to the serotonin receptor agonism by cabergoline but not bromocriptine. An approximately threefold increased prevalence of asymptomatic mild or moderate tricuspid regurgitation was detected echocardiographically in cabergoline-treated prolactinoma patients compared to controls, particularly at higher cumulative doses. Consequently, periodic cardiac exam is necessary in patients treated with cabergoline, and echocardiography is indicated if a murmur is appreciated or if high doses are required.
Surgery and radiotherapy are reserved primarily for rare tumors unresponsive to dopamine agonists and for patients intolerant to these drugs. Surgery is most effective for microadenomas, with success rates approaching 90% for microadenomas but only about 60% for macroprolactinomas. An immediate postoperative prolactin of less than 2 ng/mL is reliable evidence of cure. Radiotherapy takes at least a year to lower the prolactin significantly and to stop tumor growth. Resumption of ovulatory cycles and spontaneous conception has been reported after Gamma Knife radiosurgery.
Occasionally, patients present with normoprolactinemic galactorrhea and regular menses. If the galactorrhea is bothersome, treatment with bromocriptine or cabergoline to lower the prolactin to less than 2 ng/mL is effective in stopping the galactorrhea. The duration of therapy required is roughly proportional to the duration of time that the galactorrhea has been present. Patients should be counseled to wear a tight bra or breast binder and to avoid both nipple stimulation and testing themselves for expressible milk production during the course of therapy.
In men, the majority of patients with prolactinoma come to medical attention with macroprolactinomas and markedly elevated prolactin values. Men present with symptoms attributable either to mass effect, such as vision loss and diplopia, or to hypogonadism, including fatigue, loss of libido, and erectile dysfunction. Galactorrhea is rare but seen if gynecomastia is present and hyperprolactinemia is severe. Sperm count is usually reduced only after many years of hyperprolactinemia. Indications for treatment include mass effects and hypopituitarism. The hypogonadism associated with prolactinomas in men often responds well to dopamine agonist therapy, unless the duration of hypogonadism is prolonged. The erectile dysfunction of hyperprolactinemia does not always improve with testosterone replacement unless the prolactin is normalized. Sperm count is not immediately restored by dopamine agonist therapy and may not return to normal after many months of therapy.
Acromegaly
Acromegaly results from overproduction of GH and IGF1 accompanied by acral bone and soft tissue growth. Because symptoms are subtle and gradual in onset, the diagnosis may be delayed for several years. Additional symptoms and complications include fatigue, sleep apnea, hyperhidrosis, headache, and carpal tunnel syndrome. The majority of patients with acromegaly have a GH-secreting pituitary tumor (somatotropinomas), less than 10% have GHRH-producing tumors, usually pancreatic neuroendocrine tumors. Menstrual abnormalities are observed when tumor mass effect impairs delivery of hypothalamic releasing factors to the anterior pituitary or when hyperprolactinemia occurs. The direct action of GH can cause galactorrhea; however, tumors that cosecrete GH and prolactin also exist, which complicates the evaluation. Prolactin cosecretion does not predict an improved response to dopamine agonist therapy.
A serum IGF1, corrected for gender and age or Tanner stage, is the best screening test for acromegaly. An elevated IGF1 plus acral changes in a patient with a pituitary tumor are usually sufficient to make the diagnosis. Formal GH suppression testing with 100 grams of glucose (normal GH <0.1 ng/mL in males or <1 ng/mL in females ) is used mainly to gauge response to therapy when IGF1 is equivocal, using a value of <0.4 or <1 ng/mL as remission. Gigantism occurs when the tumor forms prior to closure of the epiphyses. Genetic causes of acromegaly and gigantism include multiple endocrine neoplasia type 1 (MEN1, MEN1 gene), Carney complex ( PRKA1A gene), and familial isolated pituitary adenoma (FIPA, AIP gene).
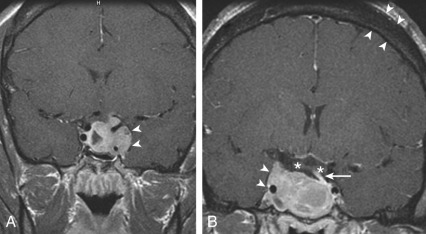
Combined modality therapy is the norm for the typical invasive somatotropinoma ( Fig. 24.4 ). Large tumors often require both transsphenoidal surgery and sometimes craniotomy to debulk the tumor sufficiently for drug or radiotherapy, whereas for microadenomas, cure rates are high and hypopituitarism infrequent for experienced pituitary neurosurgeons. Pure somatotropinomas occasionally respond to high doses of dopamine agonists with reduced GH secretion and/or tumor shrinkage. Many somatotropinomas express SS receptors, primarily type 2 (sst2), and remain responsive to SS agonists such as octreotide or lanreotide, given as monthly long-acting intramuscular or deep subcutaneous injections of 10 to 40 mg or 60 to 120 mg, respectively. Although earlier reports suggested GH and IGF1 normalization in 70% of patients, more recent studies indicate that less than 40% of patients normalize on SS agonist therapy. Some tumor shrinkage occurs in over half of somatotropinomas treated with SS analogues, but the degree of regression is not as dramatic as for prolactinomas treated with dopamine agonists. Pegvisomant, a GH receptor antagonist that is modified with polyethylene glycol, normalizes IGF1 in up to 95% of patients during the initial trials, but long-term studies indicate that the success rate is 60% to 65%. The drug is given by subcutaneous injection of 40 mg as loading dose, followed by 10 to 30 mg/day, and it is generally well tolerated except for transaminase elevation in rare cases. Pasireotide, which binds to sst5 with higher affinity than octreotide, was found to achieve biochemical control in more patients than octreotide or lanreotide but causes more hyperglycemia. GH normally rises in pregnancy due to the secretion of placental GH, which is the product of a separate gene from pituitary GH. Consequently, medical treatment is normally withheld during pregnancy.
Cushing Disease
Hypercortisolism from ACTH-producing pituitary tumors causes infertility both from the effects of glucocorticoids on the hypothalamic-pituitary-gonadal axis and from mass effect if it is caused by a macroadenoma. This topic will be covered in the adrenal section below, and the principles of hypopituitarism from macroadenomas are the same as discussed earlier.
Other Macroadenomas
Many pituitary adenomas are nonfunctional, meaning that they do not produce significant amounts of biologically active hormones. Most of these tumors, however, derive from the glycoprotein hormone cell lineage and express mRNA for the common alpha subunit and/or the beta subunits of LH, FSH, or TSH, but the glycoprotein products are biologically inactive due to improper glycosylation, dimerization, and assembly. In general, these tumors present with symptoms due to mass effect (vision loss, headache) and/or hypopituitarism. Unlike prolactinomas, these tumors tend to be resistant to medical therapy. Indications for surgery include vision compromise or other mass effect and severe hypopituitarism, such as ACTH deficiency or panhypopituitarism. For the woman with panhypopituitarism and infertility, ovulation induction with gonadotropins is required, as discussed in other chapters. For men, fertility is restored with human chorionic gonadotropins (hCG) 1000 to 2000 units 2 to 3 times weekly to normalize testosterone, with recombinant FSH 25 to 75 IU 3 times a week added if necessary.
Lymphocytic Hypophysitis
Lymphocytic infiltration of the pituitary is a rare disorder, which most commonly occurs in postpartum women. Common presentations are headache with inability to lactate postpartum due to impaired prolactin production or polyuria and polydipsia from diabetes insipidus. Some women are relatively asymptomatic but develop amenorrhea or nonspecific symptoms of hypopituitarism over time. The pattern of pituitary deficiencies is highly variable and often follows a pattern much different from that observed with macroadenomas, such as ACTH and TSH deficiency with diabetes insipidus but preserved GH and gonadotropins. During the active phase, MRI shows a symmetrically enlarged pituitary and stalk that enhances markedly with gadolinium. After the damage has occurred and the disease has subsided, the MRI can show a partial empty sella. Methylprednisolone in the acute phase is sometimes effective, but pituitary function when lost rarely recovers.
The immune-checkpoint inhibitor ipilimumab, a monoclonal antibody to the cytotoxic T lymphocyte antigen 4 (anti-CTLA-4), incites the development of lymphocytic hypophysitis in 10% to 15% of patients, with a male predominance. In contrast, nivolumab and other monoclonal antibodies to programmed cell death protein-1 (PD-1) only cause hypophysitis in 1% of cases. Headache and fatigue are common presenting features, and symmetrical pituitary enlargement is seen on MRI. TSH deficiency is the most common endocrinopathy, and primary thyroid dysfunction also occurs in up to 10% of patients ; consequently, both TSH and free T4 should be monitored in patients receiving ipilimumab. Secondary adrenal insufficiency and hypogonadism are also common but not GH deficiency. Although these patients are usually not pursuing pregnancy during treatment, ipilimumab can induce long-term cancer remission, and hypophysitis results in chronic endocrinopathies in the survivors.
Other Disorders Affecting the Pituitary
Granulomatous diseases such as sarcoidosis and tuberculosis can involve the hypothalamus and pituitary, also causing central hypogonadism. Hypopituitarism can be the initial manifestation of sarcoidosis, while others have neurosarcoidosis with manifestations such as optic neuritis. MRI shows brisk enhancement of the meninges with or without hypothalamic and pituitary abnormalities. Other tumors also arise in the hypothalamus and pituitary, including germinomas and lymphomas. Carcinomas metastasize to the pituitary, primarily to the stalk. Diabetes insipidus is often a presenting manifestation, and hypogonadism is one of the most frequent manifestations of hypopituitarism in these neoplastic disorders.
Hemochromatosis (primary or secondary) and amyloidosis are infiltrative diseases that cause hypopituitarism. The classic triad includes liver dysfunction, skin bronzing, and diabetes mellitus (“bronze diabetes”), but arthropathy, cardiomyopathy, and various endocrinopathies including adrenal insufficiency and hypogonadotropic hypogonadism are common as well. Iron deposition in the hypothalamus has predilection for impairing GnRH production among the releasing hormones. Hemochromatosis is one of the few disorders in which the male reproductive axis is more vulnerable to disruption than in the female, due to monthly menses in women. Some causes of hypopituitarism are geographical curiosities, such as hemorrhagic hypopituitarism following viper bites in Southeast Asia. Genetic causes of hypopituitarism are reviewed in Chapter 17 .
Adrenal Disorders
- ◆
Specific features of Cushing syndrome include proximal myopathy, nonblanching purple striae, dermal atrophy, and disproportionate fat accumulation in the head and neck.
- ◆
Glucocorticoid therapy for women with classic 21-hydroxylase deficiency (21OHD) seeking pregnancy should be titrated to achieve a follicular-phase progesterone <0.6 ng/mL (2 nmol/L).
- ◆
A basal or cosyntropin-stimulated 17-hydroxyprogesterone (17OHP) >1000 ng/dL (30 nmol/L) or CYP21A2 genotyping reliably diagnose nonclassic 21OHD among women with hirsutism and oligomenorrhea.
Overview
The adrenal consists of a cortex, with three distinct zones, and a medulla. The medulla is an extension of the sympathetic nervous system and produces epinephrine. The steroid-producing cells of the cortex are arranged into the outermost zona glomerulosa, which produces aldosterone; the zona fasciculata, which produces cortisol; and the zona reticularis, which produces the androgen precursor DHEAS. DHEA and DHEAS are metabolized to testosterone in peripheral tissues, and the adrenal produces small amounts of androstenedione and testosterone. The adrenal also produces rather large amounts of 11β-hydroxyandrostenedione, primarily via the 11β-hydroxylation of androstenedione, which is metabolized to the potent androgens 11β-hydroxytestosterone and 11-ketotestosterone. ACTH is the primary regulator of cortisol and DHEAS production (see Fig. 24.1 ), whereas aldosterone synthesis is mainly stimulated by the renin-angiotensin system and by potassium.
Diseases of the adrenal gland causing hormone deficiency rarely interfere with reproduction directly, but certain hormone excess states contribute to infertility, particularly for women. In the male, the contribution of adrenal DHEAS and other 19-carbon steroids to circulating testosterone concentrations is normally small, but in women, a variable amount of testosterone and all 11-ketotestosterone normally derives directly from the adrenal or from adrenal-derived precursors. Consequently, disorders that increase adrenal DHEAS production cause hyperandrogenemia in women, which can impair fertility. Hypercortisolism may suppress gonadotropin production in women and, to a lesser extent, in men. We will not discuss primary aldosteronism, adrenal insufficiency, or pheochromocytomas in this chapter because these disorders rarely impair fertility in isolation.
Adrenal Disorders That Affect Reproduction
Cushing Syndrome
The Cushing syndrome is categorized as iatrogenic or endogenous, and endogenous Cushing syndrome is dichotomized as being ACTH-dependent or ACTH-independent ( Table 24.2 ). The majority of Cushing syndrome is ACTH-dependent, the most common cause being ACTH-producing pituitary tumors, which is called Cushing disease. The differential diagnosis of ACTH-dependent Cushing syndrome also includes ectopic production of ACTH or CRH by neuroendocrine carcinomas, most commonly small cell lung cancers, or by foregut neuroendocrine tumors, formerly called “carcinoid” tumors of the thymus, bronchus, or pancreas, as well as pheochromocytomas and medullary thyroid cancers. ACTH-independent Cushing syndrome is caused by unilateral adenomas or carcinomas and by bilateral micronodular or macronodular hyperplasia. Cushing disease occurs most commonly in young women, and DHEAS production is more commonly elevated in ACTH-dependent forms of Cushing syndrome than in ACTH-independent forms. Consequently, Cushing disease in women is the most relevant form of hypercortisolism that impairs reproduction.
| ACTH-independent | Adrenocortical adenoma Adrenocortical carcinoma Macronodular hyperplasia Micronodular hyperplasia |
| ACTH-dependent | Corticotrope tumor (Cushing disease) Corticotrope hyperplasia Ectopic ACTH syndrome Ectopic CRH syndrome |
The clinical manifestations of cortisol excess can be subtle in the early stage when the diagnosis is most difficult but the benefits of treatment and potential for full recovery the greatest. Cortisol is a catabolic hormone that causes lipolysis and fat redistribution, as well as breakdown of body tissues such as muscle, skin, and bone. Central obesity is a prominent and common feature, with disproportionate fat accumulation around the face and neck. The dorsocervical fat pad (“buffalo hump”) is a well-known feature of Cushing syndrome, but supraclavicular fat pads are more specific for this disorder, especially in an otherwise nonobese individual ( Fig. 24.5 ). Facial and upper chest plethora is observed (see Fig. 24.5 ), and women often develop hirsutism.
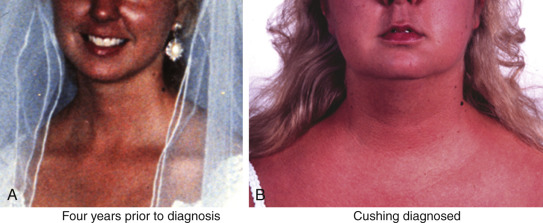
Proximal muscle weakness and skin thinning are two very specific features of Cushing syndrome, and osteoporosis in an obese individual should raise suspicion as well. Patients endorse difficulty arising from a chair and walking up stairs or getting out of a car, combing their hair, or changing an overhead light bulb. Skin thinning and capillary fragility cause easy bruising, and if weight gain is rapid, the striae formed are violaceous and nonblanching, reflecting hemorrhage into the new skin by fragile blood vessels. These striae are found on the abdomen and flanks, near the axillae, and on the thighs and breasts. If greater than 1 cm, purple striae are very specific for hypercortisolism; however, these findings develop late and only in reproductive-age women with severe disease.
The most important scenario in which Cushing syndrome should be considered is in the young woman with oligomenorrhea and hirsutism, who could be mistaken for simple polycystic ovary syndrome (PCOS). A history of hirsutism onset after age 25 or specific findings for hypercortisolism, such as easy bruising, thin skin, proximal muscle weakness, and osteoporosis, should prompt screening for Cushing syndrome.
The diagnosis of hypercortisolism can be difficult early in the disease because of the large overlap with normals. Testing is based on the principles that the cortisol production rate is increased, that the normal diurnal rhythm is disrupted, and that cortisol is not suppressible. These principles are employed by the 24-hour urinary free cortisol, late-night saliva or serum cortisol measurements, and overnight dexamethasone suppression testing, respectively. The caveats of testing are beyond the scope of the chapter, but the reader should be aware that both false-positive and false-negative results are common. Consequently, tests often must be repeated several times before a diagnosis can be made or excluded.
Once hypercortisolism is confirmed, an ACTH measurement will determine if the disease is ACTH-dependent or ACTH-independent. If ACTH-independent (ACTH <5 pg/mL), an abdominal computed tomography scan of the adrenal glands is obtained. Patients with early ACTH-independent Cushing syndrome will have low but not suppressed ACTH (5 to 15 pg/mL), and these patients should have serial testing until the diagnosis is clarified. If the ACTH is normal or elevated, the next step is MRI of the pituitary with gadolinium contrast. The caveat of pituitary imaging is that small abnormalities of the pituitary are common, and about half of patients with Cushing disease lack a visible tumor by MRI. To conclusively exclude the ectopic ACTH syndrome, inferior petrosal sinus sampling is performed. Blood draining both sides of the pituitary is sampled from the inferior petrosal sinuses under ovine CRH stimulation (100 µg bolus), and the ACTH values in these specimens are compared with those obtained in peripheral blood drawn simultaneously. An ACTH step-up greater than three from peripheral blood to the inferior petrosal sinus is reliable evidence for a pituitary source of ACTH. Neuroendocrine tumors causing ectopic ACTH production are usually identified with cross-sectional imaging of the chest, abdomen, and pelvis, or with SS-receptor scintigraphy. The [ 68 Ga]-DOTATATE or -DOTATOC PET scans are more sensitive than [ 111 In]-pentetreotide SPECT studies, but occult tumors are rarely identified with scintigraphy when cross-sectional imaging is negative.
Treatment of Cushing syndrome is primarily surgical. Cushing disease is treated with transsphenoidal pituitary adenomectomy, yet even in the hands of experienced surgeons, long-term cure rates are not above 80%. Repeat surgery, radiotherapy, and even bilateral adrenalectomy may be recommended if disease persists. Medical management with metyrapone, ketoconazole, and trilostane has been employed but are seldom very effective in severe disease. Mifepristone at high doses blocks the glucocorticoid receptor and improves the catabolic and metabolic features of all forms of Cushing syndrome, but the renal and hemodynamic manifestations of hypertension and hypokalemia must be managed independently. Cabergoline is effective in reducing hypercortisolism for a small subset of pituitary tumors that express dopamine type 2 receptors. Furthermore, many corticotrope tumors express sst5 but not sst2, so while octreotide is rarely effective in reducing ACTH secretion, pasireotide reduces ACTH and cortisol production in the majority of cases. Apparently cured patients with Cushing disease can have recurrences many years later, and screening with late-night saliva cortisol is the most sensitive test to identify biochemical recurrence. In contrast, ACTH-independent Cushing syndrome is normally cured by unilateral or bilateral adrenalectomy, except in the case of adrenocortical carcinomas with inoperable or metastatic disease.
Congenital Adrenal Hyperplasia: 21-Hydroxylase Deficiency
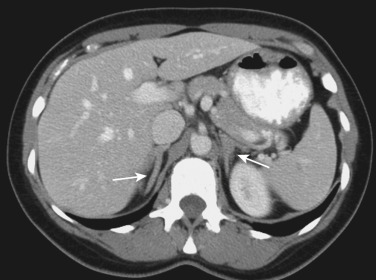
The manifestations of 21OHD in the infant as an intersex disorder were discussed elsewhere (see Chapters 4 , 16 , and 17 ), so this discussion will be restricted to adults with classic and nonclassic forms of 21OHD. Among the forms of CAH, 21OHD is by far the most common form, accounting for greater than 90% of cases. Classic 21OHD is caused by mutations in the CYP21A2 gene, which severely impair the activity of the encoded P450 21A2 enzyme to less than 2% of wild-type activity. Nearly all of these patients have clinical manifestations and require therapy with glucocorticoid and mineralocorticoid replacement. Milder mutations, particularly the V281L allele, cause nonclassic 21OHD ( Table 24.3 ). Males with nonclassic 21OHD are rarely ascertained unless evaluation for premature pubertal features (body hair, acne, growth spurt) is sought. Females with nonclassic 21OHD present with hyperandrogenism, which manifests with hirsutism, acne, and irregular menses, but not all patients are symptomatic or require treatment. Similarly, patients with intermediate severity are often not diagnosed until adulthood, particularly males ( Fig. 24.6 ). In many countries, 21OHD testing is part of newborn screening procedures, so most newborns are identified before adrenal crisis occurs, even if not ascertained by genital ambiguity.
| Form | Common Mutations |
|---|---|
| Classic | |
| Salt-wasting | Large deletions, 656A/C-G, * G110del8nt, Q318XR356W, * R483P, I236N+V237E+M239K |
| Simple virilizing † | 656A/C-G, * I172N, R356W * |
| Nonclassic | P30L, V281L, R339H, P453S |
* Can be associated with either salt-wasting or simple virilizing disease.
† All patients with classic 21-hydroxylase deficiency are prone to salt wasting with physiologic stress, but the most severely affected experience spontaneous salt-wasting crises in infancy.
The diagnosis of 21OHD is based on elevated circulating concentrations of 21-deoxysteroids, in particular 17OHP. A random serum 17OHP greater than 10,000 ng/dL with serum cortisol less than 5 µg/dL establishes the diagnosis of classic 21OHD in males and females, and dynamic testing is rarely required. In the case of nonclassic disease, a morning serum 17OHP more than 200 ng/dL should prompt repeat measurement of cortisol and 17OHP 30 to 60 minutes after cosyntropin administration (250 mcg intravenous or intramuscular). Post-cosyntropin 17OHP values in the 1000 to 10,000 ng/dL range are typical of nonclassic 21OHD. If laboratory data are equivocal, genetic testing for mutations in the CYP21A2 gene on DNA from peripheral blood cells is available.
The repertoire of mutations commonly found in 21OHD is limited due to the molecular mechanism responsible for most cases. The CYP21A2 gene is located in a duplicated locus within the HLA region on chromosome 6p that includes the genes for the fourth component of complement. In the duplicated region, the DNA corresponding to the CYP21A2 gene is replaced by the CYP21A1P pseudogene. This pseudogene contains several mutations that render the cognate mRNA and protein nonfunctional. Most cases of 21OHD derive from gene conversion events, in which some or all of the CYP21A2 gene is replaced by the corresponding region of the CYP21A1P pseudogene. Consequently, the spectrum of mutations and the worldwide prevalence of 21OHD is fairly consistent, but certain mutations are particularly common in specific populations, such as the intron 2 A-to-G mutation in Yupik Eskimos, which accounts for the high prevalence of classic 21OHD in that population. Occasionally, true point mutations are found instead. Heterozygous carriers are identified with confidence by genetic testing, whereas 17OHP values, even after cosyntropin stimulation, show broad overlap with normal individuals of either sex.
The mechanisms of reduced fertility in women with classic 21OHD are complex, although affected women have given birth to normal female infants. Chronic hyperandrogenemia, even though largely adrenal in origin, contributes to chronic oligo-anovulation. In addition, many women with 21OHD develop a secondary PCOS, with characteristic ovarian morphology, thecal hyperplasia, and ovarian androgen excess as well. The block in 17OHP metabolism causes accumulation of adrenal-derived progesterone upstream of the enzymatic defect, and high circulating progesterone concentrations impair endometrial maturation and implantation. Overtreatment with synthetic glucocorticoids, however, can suppress gonadotropins and cause glucose intolerance with its attendant reproductive disturbances.
Women with virilization of the external genitalia face additional impediments to fertility related to anatomical considerations. Vaginal stenosis, either prior to reconstruction surgery or following suboptimal surgery and/or dilation, is known to cause dyspareunia, which discourages coitus. Vaginal anomalies also compromise sperm deposition, and high circulating progesterone concentrations inhibit sperm penetration and fertilization. Psychosocial influences should not be underestimated. The influence of prenatal and/or early androgen exposure, the psychologic adjustment to masculinization, and the presence of sexual dysfunction might all be significant factors contributing to gender identity and sexual preference women with CAH.
Women with nonclassic 21OHD are most commonly identified during an evaluation of hirsutism, oligomenorrhea, or infertility mimicking PCOS. It is difficult to distinguish nonclassic 21OHD from idiopathic PCOS on clinical grounds ; therefore, guidelines suggest screening for nonclassic 21OHD with a 17OHP before making the diagnosis of PCOS. More careful studies suggest that the frequency of infertility or chronic anovulation in women with nonclassic 21OHD is not much higher than in the general population. Nevertheless, if infertility and/or chronic anovulation are present in a woman with nonclassic 21OHD, these women often benefit from glucocorticoid replacement when trying to conceive. Curiously, about 70% of women with nonclassic 21OHD in one large series are compound heterozygotes for one nonclassic and one classic allele, whereas the majority should be homozygous for nonclassic alleles given population estimates of carrier frequencies. This result suggests that the less severely affected nonclassic 21OHD patients rarely come to medical attention. Furthermore, cryptic and largely asymptomatic nonclassic 21OHD was found in 4% of parents with children affected by classic 21OHD, all of whom were heterozygous carriers of classic alleles.
Little data exist to guide the optimal management of adults with 21OHD, either for long-term glucocorticoid replacement or for improving fertility. A range of treatment regimens are used without obvious rationale, although the convenience of once-daily dosing has favored use of the more potent glucocorticoids including prednisone, prednisolone, and dexamethasone. In a large audit of 21OHD patients in England, many were short and obese, with poor health metrics and quality of life, and overtreatment with glucocorticoids appeared to be common ; a natural history study at the National Institutes of Health revealed similar findings. Consequently, the minimum amount of glucocorticoid replacement to maintain testosterone in an acceptable range is recommended for chronic therapy in women, although this dose and target steroid values will vary among individuals. Serum 17OHP should not be normalized, as occurs with over-treatment, but dose adjustments should be based on serum testosterone and androstenedione values. Hydrocortisone is preferable for chronic therapy but must be given in at least two divided doses, although sustained-release preparations are in development, and continuous subcutaneous infusion is very effective but labor-intensive and off-label. Prednisolone and dexamethasone lower androgen production more effectively than hydrocortisone, particularly for the morning rise in ACTH and adrenal-derived steroids, where even bedtime hydrocortisone is not very effective. For classic 21OHD patients, fludrocortisone acetate in doses sufficient to normalize plasma renin activity and serum potassium, eliminate orthostasis, and maintain fluid balance, should be maintained throughout life with rare exceptions.
Despite the many barriers to achieving pregnancy in women with 21OHD, fecundity rates in those women who attempt pregnancy are greater than 90%, equivalent to the general population. The most critical parameter favoring conception appears to be follicular-phase serum progesterone, which should be lowered to less than 0.6 ng/mL (2 nmol/L) with multiple doses of hydrocortisone and/or prednisolone. A bedtime dose of 0.5 to 2 mg prednisolone, which is combined with either daytime hydrocortisone or prednisolone (one or two doses), appears to be particularly important for achieving pregnancy. Ovulation induction with standard methods and agents might be necessary despite optimal adrenal replacement regimens. Despite clinical features of PCOS shared with 21OHD, metformin and other insulin sensitizers have not been studied adequately in women with 21OHD. Prenatal treatment with dexamethasone has been shown to reduce fetal virilization in couples who have one or more children with 21OHD, but this treatment is considered experimental due to the paucity of long-term outcomes data in treated children with or without 21OHD.
Almost all men with nonclassic 21OHD are asymptomatic and can be identified only with genetic testing. Men with classic 21OHD, in contrast, frequently develop infertility, particularly if poorly controlled. Severe adrenal androgen excess causes gonadotropin suppression with subsequent Leydig cell atrophy, reduced testicular testosterone production, and impaired spermatogenesis. Glucocorticoid replacement often normalizes testicular function, although high doses are often required initially. More commonly, testicular adrenal rest tumors (TARTs) are the cause of male infertility in 21OHD. The steroid-producing cells of the adrenal cortex and the gonads derive from the same pool of precursors during embryogenesis. The migration of adrenal cells to the suprarenal space can be imperfect, and adrenal cortex cells or progenitors appear to reside in the testis or within the groin and abdomen. ACTH is trophic for these adrenal rest cells as well, and when ACTH is high as in poorly controlled 21OHD, these cells will form masses, most often in the testis itself. TARTs are present in 30% to 60% of men with 21OHD, and their size correlates better with adrenal volume than with single steroid measurements. Testicular ultrasonography is the most sensitive method of diagnosis, although many are palpable. Because the testis is confined to a rigid capsule, the growth of TARTs impairs testicular blood flow and efflux of sperm into the ejaculate.
Treatment of TARTs often requires potent glucocorticoids, such as dexamethasone, 1 to 2 mg at night, although even this therapy is not always successful in reducing tumor size and restoring fertility. Combination therapy with daytime hydrocortisone and low-dose (0.1 mg) bedtime dexamethasone also appears effective and less toxic than dexamethasone monotherapy at higher doses. Surgical resection of the TARTs by a competent urologist is associated with a low risk of recurrence but with little improvement in spermatogenesis or testosterone synthesis. This result, however, could reflect a selection bias in reserving surgery for the most severely affected men. The presence of TART and elevated serum FSH are poor prognostic factors for fertility in men with classic 21OHD.
Other Congenital Adrenal Hyperplasia
The other forms of CAH associated with androgen excess in women are 11-hydroxylase deficiency (11OHD) and 3β-hydroxysteroid dehydrogenase/isomerase deficiency (3βHSDD). In 11OHD, caused by mutations in the CYP11B1 gene that encodes P450 11B1, the 11-deoxysteroids 11-deoxycorticosterone (DOC) and 11-deoxycortisol are markedly elevated, and excess DOC causes hypertension and hypokalemia, unlike 21OHD. Serum 17OHP may be elevated in 11OHD, but much less than in 21OHD with similar hyperandrogenemia. In 3βHSDD, caused by mutations in the HSD3B2 gene, the diagnostic steroid ratios are those of delta-5 to delta-4 steroids, such as 17-hydroxypregnenolone/cortisol ratios greater than 6 standard deviations above normal. Note that circulating testosterone concentrations are paradoxically elevated in women with 3βHSDD. The delta-5 steroids DHEA and DHEAS are metabolized to active androgens in the periphery and converted to delta-4 steroids by 3βHSD type 1, which is abundant in liver and skin, explaining this phenomenon.
The mechanisms responsible for infertility in men and women with 11OHD are similar to those in 21OHD. Treatments are similar, except that mineralocorticoid receptor antagonists (spironolactone, eplerenone) are useful to control hypertension during chronic therapy. Note that spironolactone is also an androgen receptor antagonist and is contraindicated in women who have unprotected intercourse. Women with 3βHSDD, even in the most severe cases, have mild clitoromegaly and little labioscrotal fusion, so anatomical impediments to pregnancy are less important, and high progesterone is less of a negative factor for reproduction than in 21OHD and 11OHD.
Forms of CAH that impair both androgen and estrogen production include combined 17-hydroxylase/17,20-lyase deficiency, caused by mutations in the CYP17A1 gene encoding P450 17A1, and isolated 17,20-lyase deficiency, due to mutations in CYP17A1 or its redox partner proteins cytochrome P450-oxidoreductase ( POR mutation G539R) and cytochrome b 5 . Although all patients with complete 17OHD appear phenotypically as prepubertal females without treatment, those with 46,XX karyotype have chronic anovulation and as a result are prone to develop large cystic ovaries that can hemorrhage. These women can develop some secondary sexual characteristics and even cyclic menses if the enzymatic defect is incomplete. Nevertheless in vitro fertilization has been achieved with aspirated oocytes, and live triplets were reported after transfer of cryopreserved embryos in a patient with partial 17OHD. A term pregnancy in a woman with nearly complete 17OHD was described recently. Viable embryos were obtained from an initial superovulation and in vitro fertilization cycle, but she miscarried. The critical strategy during the second cycle—as also used in the case bearing live triplets—was to suppress both ovarian and adrenal-derived progesterone with GnRH agonist and dexamethasone, respectively, prior to endometrial preparation with estradiol valerate and implantation of frozen embryos. Estrogen treatment was stopped at 14 weeks, and the baby was delivered at 30 weeks.
Lipoid CAH, due to mutations in the STAR gene encoding the steroidogenic acute regulatory protein (StAR) or rarely the CYP11A1 gene encoding P450 11A1, abrogates all steroid production, although a nonclassic form with partial StAR deficiency mainly impairs cortisol production. One 46,XX woman with lipoid CAH achieved pregnancy three times at age 25 to 30 following clomiphene stimulation and progesterone support, and the second and third pregnancies resulted in live twin and singleton births, respectively. POR deficiency manifests as a spectrum of phenotypes with disordered steroidogenesis, from primary amenorrhea in phenotypically normal women to the Antley-Bixler syndrome, with skeletal dysmorphologies. Patients with POR deficiency are generally considered infertile, although most of the patients diagnosed with this condition since its description in 2004 are children. Milder cases, in which pregnancies might be more likely than in more typical disease, are also less likely to be evaluated for this diagnosis.
Related posts:
 Gonadotropin Hormones and Their Receptors
Gonadotropin Hormones and Their Receptors
 Steroid Hormones and Other Lipid Molecules Involved in Human Reproduction
Steroid Hormones and Other Lipid Molecules Involved in Human Reproduction
 Structure, Function, and Evaluation of the Female Reproductive Tract
Structure, Function, and Evaluation of the Female Reproductive Tract
 Hormone-Responsive Cancers
Hormone-Responsive Cancers
 Medical Approaches to Ovarian Stimulation for Infertility
Medical Approaches to Ovarian Stimulation for Infertility
 Pelvic Imaging in Reproductive Endocrinology
Pelvic Imaging in Reproductive Endocrinology
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


