Abstract
Physiological and endocrine adaptations occur in the mother in response to the demands of pregnancy. These demands include support of the fetus (volume support, nutritional and oxygen supply, and clearance of fetal waste), protection of the fetus (from starvation, drugs, toxins), preparation of the uterus for labor, and protection of the mother from potential cardiovascular injury at delivery. The presence of a preexisting endocrine disorder is likely to affect the ability of the mother to adapt to the demands of pregnancy and, as a result, may influence fetal growth and development. Drugs used to treat such disorders may also affect perinatal outcome. The most common preexisting endocrine disorders that can complicate pregnancy are diabetes mellitus, thyroid dysfunction, and obesity. Less common preexisting maternal endocrine disorders include pituitary tumors, diabetes insipidus, and hyperparathyroidism.
The physiological and endocrine adaptations that characterize pregnancy can also lead to the development of pregnancy-specific diseases in previously healthy women, the most common of which are gestational diabetes and disorders of the endocrine and sympathetic nervous systems associated with preeclampsia and preterm labor. This chapter is designed to review in detail the underlying pathophysiology of these pregnancy-specific diseases, as well as the effects of pregnancy on preexisting endocrine disorders. A better understanding of these conditions will improve the ability of clinicians to optimize maternal and perinatal outcome in such pregnancies.
Keywords
Diabetes mellitus, thyroid disease, obesity, hypothalamic-pituitary axis, adrenal disease, ovarian tumor, preeclampsia, parturition, preterm birth
Diabetes Mellitus
- ◆
Pregnancy is a diabetogenic state. Control of maternal glucose metabolism is shared by the mother and the fetoplacental unit. Changes such as insulin resistance and reduced peripheral glucose uptake provide a continuous supply of glucose for the developing fetus.
- ◆
The fetoplacental unit is responsible for pregnancy-induced insulin resistance, primarily through the production of antiinsulin hormones, including growth hormone, human chorionic somatomammotropins, cortisol, and progesterone.
Effects of Pregnancy on Maternal Glucose Metabolism
Normal pregnancy can be regarded as a diabetogenic (prodiabetic) state, with evidence of insulin resistance, maternal hyperinsulinism, and reduced peripheral uptake of glucose. These endocrine alterations, which result primarily from the production of antiinsulin hormones from the placenta (see Chapter 11 ), are designed to ensure a continuous supply of glucose for the developing and growing fetus. Therefore, in pregnancy, the control of maternal glucose metabolism is shared by the mother and the fetoplacental unit. The endocrine and molecular mechanisms by which the fetoplacental unit is able to reset the carbohydrate homeostatic equilibrium in the mother are not clear, but likely involve the action of several placental hormones. These hormones include growth hormone (GH), human chorionic somatomammotropins (hCS; placental lactogens), corticotropin-releasing hormone (CRH), cortisol, and progesterone.
Insulin Production and Action Changes During Pregnancy
Major functional changes occur in insulin production and action during pregnancy. β-cells in the islets of Langerhans within the pancreas—the cells responsible for insulin production—undergo hyperplasia, leading to insulin hypersecretion and an increase in circulating insulin levels throughout pregnancy. It is this mechanism, along with the hemodilution of pregnancy and the initial enhanced responsiveness of cells to circulating levels of insulin, that is likely responsible for the fasting hypoglycemia seen in early pregnancy. However, as pregnancy progresses, peripheral resistance to insulin increases. In an attempt to overcome this resistance, the pancreas further increases insulin secretion. This compensatory response serves to return circulating maternal glucose levels to the normal range, but results also in chronically elevated insulin levels (both in the fasting and fed state), in the postprandial hyperglycemia that characterizes normal pregnancy, and in islet cell hyperplasia.
Beta cell hyperplasia and expansion are controlled, in part, by prolactin (PRL) and hCS, which both cause an increase in the number of pancreatic β-cells in pregnancy.
Data from mice have provided some insights into the mechanisms regulating β-cell hyperplasia. Kim and colleagues found that serotonin acts downstream of lactogenic hormone signaling via the PRL receptor to stimulate β-cell proliferation. Karnik and colleagues. reported that repression of menin—the protein product of the MEN1 gene—is necessary for normal B-cell proliferation, and that PRL represses menin levels in pancreatic islets.
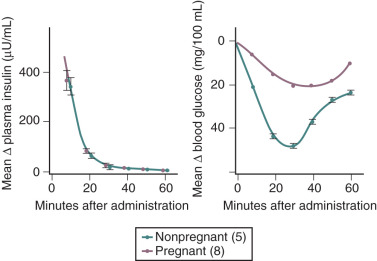
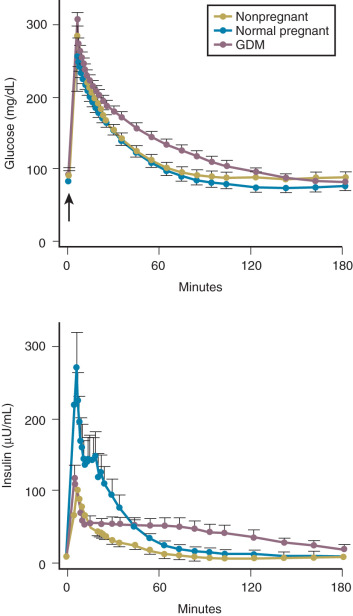
Insulin resistance refers to a decrease in the ability of a fixed concentration of circulating insulin to stimulate peripheral glucose uptake in adipocytes and muscle cells. The condition can be demonstrated either by the insulin tolerance test or by glucose loading tests. An insulin tolerance test involves the injection of a standard dose of insulin followed by serial blood glucose measurements. The clearance of insulin from the circulation is not altered by pregnancy ( Fig. 27.1 ). The half-life of insulin is approximately 7 minutes both before and during pregnancy. However, in pregnant subjects, the administration of insulin results in a smaller decline in circulating glucose than that seen in nonpregnant subjects (see Fig. 27.1 ). Furthermore, in pregnancy, intravenous ( Fig. 27.2 ) or oral ( Fig. 27.3 ) administration of glucose causes significant hyperinsulinemia as compared with the nonpregnant state, resulting in relative hyperinsulinemia after meals. Taken together, these data provide evidence in support of pregnancy being an insulin-resistant state.
The molecular mechanisms responsible for the insulin resistance in pregnancy are not well understood, but several factors are likely involved. Although insulin receptor kinase activity does not appear to be affected by pregnancy, the numbers of high-affinity insulin receptors on the surface of adipocytes are threefold lower in pregnancy than in nonpregnant women. The glucose transport system also appears to be perturbed in pregnancy, with a threefold reduction in insulin-stimulated glucose transport as compared with nonpregnant controls.
The movement of glucose into adipocytes and skeletal muscle cells is mediated by the glucose transport proteins, GLUT-1 and GLUT-4. GLUT-1 is responsible for basal glucose transport and is not responsive to insulin. Insulin increases glucose uptake in cells by stimulating the translocation of GLUT-4 from intracellular sites to the cell surface. Up to 75% of insulin-dependent glucose disposal occurs in skeletal muscle, whereas adipose tissue accounts for only a small fraction. In some pregnancies complicated by gestational diabetes, GLUT-4 is markedly reduced and fails to translocate to the cell surface with insulin stimulation, leading to a reduction in glucose transport in both the basal and insulin-stimulated states. Taken together, these data suggest that the peripheral insulin resistance that characterizes pregnancy likely results from several integrated mechanisms, including a decrease in insulin receptor number, a “postreceptor” defect in insulin action, and alterations in glucose transport systems. The postreceptor mechanisms that contribute to insulin resistance in late pregnancy occur in skeletal muscle at the β-subunit of the insulin receptor, at the level of insulin receptor substrate-1 (IRS-1), and in the cytoplasm, where increased free intracytoplasmic p85α regulatory subunit of phosphatidylinositol 3-kinase leads to decreased ability of insulin to stimulate the association of catalytic proteins with IRS-1. Together these alterations in insulin signaling likely result in less glucose uptake in skeletal muscle.
Impaired suppression of nonesterified fatty acids (NEFAs) in pregnant women suggests another mechanism for insulin resistance in pregnancy. NEFAs have been demonstrated to impair insulin-stimulated hepatic glucose uptake and whole body glucose disposal. Impaired NEFA suppression to endogenous insulin has been observed in a pregnant population subjected to a hyperinsulinemic clamp. Thus inappropriately elevated NEFA levels may play a role in the insulin resistance observed in pregnancy. Dysregulation of NEFA has been implicated in both insulin resistance in normal pregnancy and decreased insulin secretion in gestational diabetes, and has been posited as a pathophysiological link between gestational diabetes and the subsequent development of type 2 diabetes (DM2).
Fetoplacental Counterregulatory Hormones
Although the molecular mechanisms have yet to be fully elucidated, the fetoplacental unit is clearly responsible for the pregnancy-induced insulin resistance, exerting its effect largely through the production of counterregulatory (antiinsulin) hormones. Insulin promotes the uptake of glucose by adipocytes and muscle cells. Counterregulatory hormones inhibit insulin-mediated glucose uptake by adipocytes and muscle cells, acting largely at a postreceptor level. Such hormones include, among others, GH, hCS, cortisol, and progesterone.
Placental Growth Hormone and Human Chorionic Somatomammotropins
Placental GH differs from pituitary GH by 13 amino acid (191 nucleotide) substitutions and circulates in at least three isoforms: 22, 24, and 26 kDa. hCS are single-chain proteins produced largely by the syncytiotrophoblast with a high degree of sequence homology to both GH and PRL . In primates, hCS genes appear to have evolved from a precursor GH gene; in nonprimate species, on the other hand, the placental lactogens appear to have evolved from a precursor PRL gene. Because of these differences in evolution, we have chosen to use the term “human chorionic somatomammotropins” to refer to these genes collectively. The dominant isoform of hCS is 191 amino acids in length, with a molecular mass of 23 kDa. hCS binds with high affinity to PRL receptors, but with low affinity to GH receptors, suggesting that it functions largely as a lactogen rather than as a somatogen in pregnancy. Conversely, placental GH binds with high affinity to GH receptors but with low affinity to PRL receptors. Thus, by midgestation, the endocrinological milieu is one of high levels of two lactogenic hormones (PRL and hCS), and placental GH, which is acting almost exclusively as a somatogen. hCS is also secreted directly into the fetal circulation, but is present in much lower levels than fetal PRL. Placental GH, however, is detectable only in maternal blood.
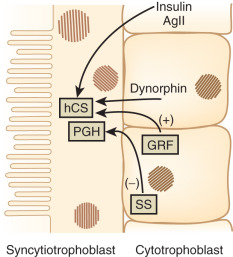
The factors that regulate GH and hCS synthesis and secretion are not fully understood, but somatostatin and GH-releasing hormone produced by the cytotrophoblast may play inhibitory and stimulatory roles, respectively. Additional regulation of hCS may be provided by insulin and angiotensin II, which both stimulate hCS release, as well as by dynorphin ( Fig. 27.4 ). This placental endocrine and paracrine-autocrine regulatory system is similar to that observed in the hypothalamic-pituitary axis, which has led Dr. Samuel Yen to refer to the placenta as “the third brain.”
The genes coding for GH and hCS are clustered together in a single region of chromosome 17 in the following order (5′ to 3′): hGH-N (pituitary GH gene), hCS L , hCS-A , hGH-V (placental GH gene), and hCS-B. The pattern of expression of these genes is tissue-specific and changes throughout gestation. For example, the placenta does not express the hGH-N (pituitary GH) gene.
Pituitary GH is secreted in a pulsatile fashion from the maternal anterior pituitary, and can be measured in the maternal serum throughout the first trimester of pregnancy. Thereafter, however, pituitary GH secretion progressively declines. By the third trimester of pregnancy, pituitary GH secretion is effectively suppressed and cannot be rescued by the induction of a hypoglycemic stimulus or by amino acid infusions (which will be discussed later in this chapter). In contrast, circulating concentrations of placental GH—encoded by the hGH-V (placental GH) gene and expressed exclusively in the placenta—increase progressively throughout the second and third trimesters of pregnancy. Similar differences are seen in the expression of the hCS genes. For example, at 8 weeks’ gestation, the hCS-A and hCS-B genes are expressed equally in the placenta; however, at term, expression of hCS-A is 5 times greater than that of hCS-B. Results of radioreceptor assay studies suggest that the relative contributions to circulating GH-like activity in pregnancy at term are 85% from hGH-V (placental GH), 12% from hCS, and less than 3% from hGH-N (pituitary GH). Multiple genes, multiple mRNA species (as in the hCS-L and hGH-V genes, which generate two distinct mRNA transcripts on the basis of alternative splice-acceptor sites for each gene), and heterogeneity in posttranslational processing result in many isoforms of these key placental hormones. The potential teleological advantages of having multiple placental GH-like genes are to ensure that the placenta can generate sufficient quantities of GH-like hormone to regulate maternal and fetal metabolism, and to minimize the risk of pregnancy failure due to a functional “knockout” of any single gene.
During pregnancy there is a major transition in the locus of control of the GH axis from the maternal hypothalamic-pituitary unit to the placenta. Circulating levels of placental GH and hCS increase throughout pregnancy. These proteins act through cell surface receptors; GH and PRL receptors belong to a superfamily of cytokine receptors that share a high degree of sequence homology to stimulate the production of insulin-like growth factor-1 (IGF-1). Circulating concentrations of IGF-1 in the maternal serum increase throughout pregnancy, reaching a peak near term. This increase in IGF-1 has also been observed in a pregnant dwarf with complete pituitary GH deficiency, suggesting that placental hormones may mediate this effect. Concentrations of IGF-binding protein-1 (IGFBP-1) rise in the first trimester, reach a peak at approximately 12 to 14 weeks of gestation, and thereafter remain stable for the remainder of pregnancy. The level of unbound (bioavailable) IGF-1 therefore increases as pregnancy progresses, and likely contributes to the suppression of hGH-N (pituitary GH) gene expression in the latter half of gestation.
In the circulation, GH can exist in a free form or bound to a GH-binding protein (GHBP). Approximately 30% of circulating GH is bound to GHBP and therefore not biologically active. Veldhuis and colleagues proposed that GHBP may serve as a buffer to prevent the level of free (bioavailable) GH from falling too low between secretory pulses. GHBP is the ectodomain of a larger cellular GH receptor and is released into the circulation after proteolytic cleavage of the parent molecule. It is likely, therefore, that circulating concentrations of GHBP parallel that of the cellular GH receptor in important target organs like the liver. This relationship allows for a balance between GH action (mediated through the GH receptor) and inactivation (by binding to the GHBP). The greater the levels of circulating GHBP, the greater the concentration of cellular GH receptors and the greater the sensitivity of cells to the actions of GH. GHBP concentrations tend to decline as gestation advances, although the reason for this change and its physiological significance remain unclear.
This system is not a prerequisite for pregnancy success, because a normal pregnancy is possible in women with Laron dwarfism, in which both the GH receptor and GHBP are absent. However, aberrations in this system may be associated with pregnancy-related complications. For example, GHBP levels have been shown to be significantly higher in women with gestational diabetes compared with nondiabetic pregnant women. This observation suggests that the concentration of GH receptors is also increased in women with gestational diabetes, leading to a degree of sensitization to the effects of GH and hCS. An increased sensitivity to the effects of circulating GH could explain many of the endocrine changes observed in gestational diabetes, including insulin resistance, higher serum glucose levels, and an increased incidence of fetal macrosomia.
Differential levels of placental GH and hCS have been noted in normal and pathological pregnancies. Low maternal levels of placental GH and hCS have been noted in pregnancies complicated by hypertension, preeclampsia, and intrauterine growth restriction (IUGR). Conversely, high levels of hCS have been noted in the blood of pregnant mothers with gestational diabetes.
Placental gene expression profiling has demonstrated different mRNA expression profiles of placental GH and hCS in pregnancies affected by preeclampsia and GDM, compared with normal pregnancies. The same investigators also demonstrated downregulation of the entire GH/hCS cluster in the placenta in pregnancies with small-for-gestational-age newborns, with significantly increased expression of hCS mRNA placental transcripts in pregnancies resulting in large-for-gestational-age newborns.
Uteroplacental insufficiency, as seen in preeclampsia and other maternal hypertensive disorders, may lead directly to decreased placental GH expression, resulting in decreased maternal lipolysis and decreased maternal IGF-I.
Circulating levels of placental GH (hGH-V), IGF-1, and the IGFBPs appear to correlate with birth weight. For example, a decrease in both placental GH and IGF-1 levels has been associated with IUGR. The decrease in placental GH levels is due to both a decrease in placental mass and a decrease in the density of placental GH-secreting cells. There is also a strong inverse correlation between maternal IGFBP-1 concentrations and birth weight in both term and preterm pregnancies. The higher the IGFBP-1 concentration, the lower the circulating level of unbound (bioavailable) IGF-1 and the lower the birth weight. Moreover, several investigators have reported a positive correlation between birth weight and circulating concentrations of IGF-1 in the fetus and neonate.
Reece and colleagues reported that IGF-1 concentrations were significantly lower in neonates below the mean birth weight for gestational age than levels in neonates above the mean (mean ± SEM: 40 ± 11 vs. 86 ± 6 ng/mL, respectively), with no differences in IGF-2 concentrations. Similar findings were reported by Lassarre and colleagues.
Conversely, maternal hyperglycemia may increase placental and fetal weight via induction of IGF-2 and fetal hyperinsulinemia. Increased maternal fat stores may reduce plasma adiponectin, and thus increase hCS expression via decreased adiponectin suppression. Increased hCS in the fetal circulation may promote fetal hyperinsulinemia via induction of β-cell replication, increasing fetal weight gain. Taken together, these data suggest that GH, hCS, IGF-1, as well as their binding proteins (GHBP and the IGFBPs) may play an important role in governing fetal growth and pregnancy outcome, and that these endocrine factors may be regulated by both the mother and the fetoplacental unit.
Cortisol
Cortisol is a potent diabetogenic hormone. It promotes lipolysis in adipocytes and protein breakdown in muscle, leading to an increase in circulating free fatty acids and amino acids. Levels of adrenocorticotropic hormone (ACTH) and cortisol increase in pregnancy. The increase in ACTH is due, at least in part, to an increase in CRH production by the placenta (discussed later in this chapter). Much of the increase in total cortisol concentration in pregnancy is due to the excessive production of corticosteroid-binding globulin by the liver under the influence of estrogen. However, there is also a significant increase in urinary free cortisol excretion during pregnancy, suggesting that circulating levels of free cortisol may also be increased. The relative contribution of an increase in circulating free cortisol to the insulin resistance of pregnancy is unclear.
Progesterone
High concentrations of progesterone have been shown to cause insulin resistance in cells in culture and in laboratory animals by decreasing the insulin receptor number and causing a postreceptor defect in insulin action, which has yet to be fully elucidated. High circulating concentrations of progesterone may contribute to pregnancy-related insulin resistance.
Fasting State
In nonpregnant women, there is a constant need to maintain circulating glucose concentrations for use by the brain. During an overnight fast, glucose is released from the liver by both glycogenolysis (breakdown of glycogen stores [75%]) and gluconeogenesis (production of glucose from circulating metabolic precursors [25%]). The precursors for gluconeogenesis include pyruvate, alanine (from muscle), glycerol (from the breakdown of triglycerides in adipose tissue), and lactate (from anaerobic metabolism).
Pregnancy is associated with an increased demand for glucose and alanine, both of which are required by the developing fetus. As such, the fasting state in pregnancy is characterized by a rapid and often severe decrease in maternal serum glucose and alanine concentrations. Associated with these changes is an increase in the circulating levels of free fatty acids (derived from triglyceride breakdown in adipose cells) and ketone bodies (see Fig. 27.3 ; Table 27.1 ). The hyperketonemia that characterizes late pregnancy is the result of enhanced lipolysis, which is likely due, in turn, to the insulin resistance in adipocytes caused primarily by the placental counterregulatory hormones.
| Measurement (Mean ± SEM) | ||
|---|---|---|
| Nonpregnant State | Late Pregnancy | |
| Glucose (mg/dL) | 79 ± 2.4 | 68 ± 1.5 * |
| Insulin (µ/mL) | 9.8 ± 1.1 | 16.2 ± 2.0 * |
| Glucagon (pg/mL) | 126 ± 6.1 | 130 ± 5.2 |
| Amino acids (µM) | 3.82 ± 0.13 | 3.18 ± 0.11 * |
| Alanine (µM) | 286 ± 15 | 225 ± 9 * |
| Free fatty acids (mg/dL) | 76 ± 7 | 181 ± 10 * |
| Cholesterol (mg/dL) | 163 ± 8.7 | 205 ± 5.7 * |
In pregnancy, the acceleration of lipid catabolism during fasting helps the mother rely on fat as a major energy source, thereby minimizing protein catabolism (preserving muscle mass) and allowing both glucose and amino acids to be used preferentially by the fetus. These metabolic adaptations have been termed “accelerated starvation” by Freinkel. Although a useful descriptive term, this characterization of pregnancy is largely inaccurate, because fat mass is known to increase significantly during pregnancy.
Fed State
The fetus is a thief! Many of the metabolic and endocrine adaptations associated with pregnancy are designed to maintain a preferential and uninterrupted supply of metabolic fuel from mother to fetus, as dictated by the progressively increasing demands of the growing fetus. The placenta is relatively impermeable to fat, but readily transports glucose, amino acids, and ketone bodies from the maternal to the fetal circulation.
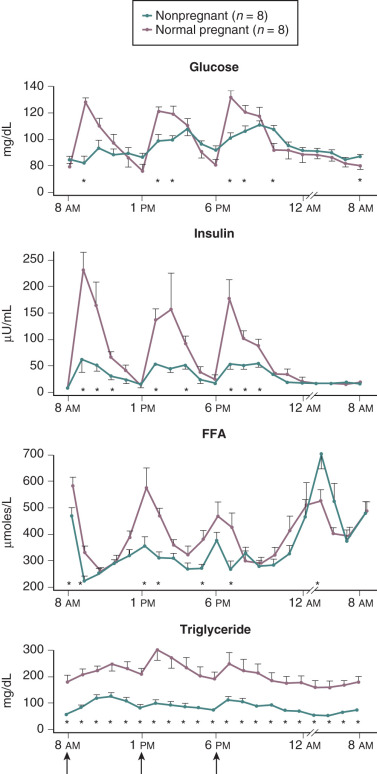
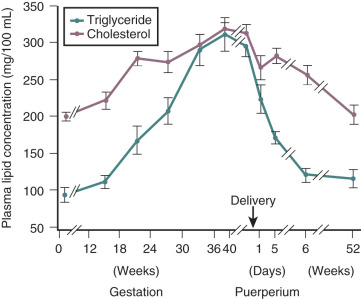
Pregnancy is associated with hyperlipidemia, both in the fasting and the fed states. Total plasma lipid concentrations increase progressively after 24 weeks of gestation. Increases in triglycerides, cholesterol, and free fatty acids are significant ( Figs. 27.5 and 27.6 ; see Table 27.1 ). High-density lipoprotein cholesterol levels rise during early pregnancy, and low-density lipoprotein cholesterol concentrations increase in later pregnancy.
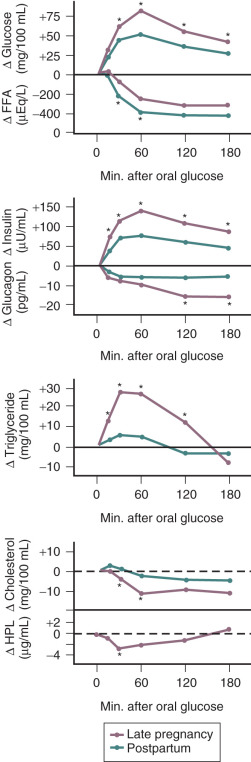
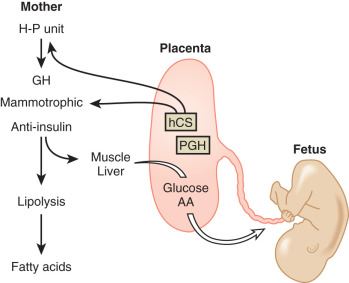
In pregnancy, an oral glucose load is associated with a greater increase in circulating glucose concentration, a smaller decline in free fatty acids, and a larger increase in serum triglycerides than that seen in the nonpregnant state (see Fig. 27.5 ). A similar effect has been observed in pregnancy after meals (see Fig. 27.3 ). These adaptations allow the mother to use primarily available triglycerides, glycerol, and free fatty acids for metabolic fuel after meals, and to preserve glucose and amino acids for preferential use by the fetus ( Fig. 27.7 ). The cause of these metabolic changes is likely the lipolytic action of the placental counterregulatory hormones (GH, hCS, cortisol, and progesterone), which serve to promote lipolysis during fasting and hypertriglyceridemia in the fed state.
Pancreatic β-Cells: The Missing Link
To a point, pancreatic β-cells will respond to insulin resistance with increased insulin secretion. Bergman and colleagues first characterized a predictable hyperbolic relationship between the quantity of insulin produced by β-cells and tissue sensitivity to insulin. The disposition index, or the “hyperbolic correction,” is a measure of insulin secretion corrected for insulin resistance. A left-shifted curve, representing decreased compensatory insulin secretion for a given level of insulin resistance, may be seen in women who develop both gestational diabetes (GDM) and type 2 diabetes mellitus (DM2). Buchanan posits that insulin resistance actually causes the β-cell dysfunction observed in GDM, and that chronic insulin resistance leading to β-cell failure may be the mechanism by which women with GDM progress to DM2.
Gestational Diabetes
- ◆
Gestational diabetes mellitus (GDM) may be difficult to distinguish from prepregnancy diabetes. There is no consensus on whether women with a positive diabetes screen in the first trimester of pregnancy should have a unique designation.
- ◆
There is also a lack of consensus on how and when to screen for gestational diabetes. Some advocate a one-step screening process (75 g, 2-hour oral glucose tolerance test [OGTT]), while others advocate two-step screening (50-g, 1-hour glucose challenge test [GCT], followed by a 100-g, 3-hour OGTT for diagnosis if the initial test is positive).
- ◆
Gestational diabetes is associated with increased maternal and fetal morbidity, including but not limited to increased risk of hypertensive disorders of pregnancy, cesarean delivery, fetal macrosomia, stillbirth, and neonatal hypoglycemia.
- ◆
The balance of evidence suggests that treating gestational diabetes to optimize glycemic control decreases the risk of preeclampsia, fetal macrosomia, and shoulder dystocia. Both oral agents (glyburide, metformin) and insulin are accepted therapies.
Depending on the patients screened and the diagnostic criteria used, GDM complicates between 6% and 20% of all pregnancies in the United States. Prevalence has been increasing over time, likely due to increases in mean maternal age and weight. The American Diabetes Association (ADA) classification of diabetes mellitus is summarized in Box 27.1 . GDM has previously been defined as any degree of carbohydrate intolerance with the onset of pregnancy or first recognized during pregnancy. The American Congress of Obstetricians and Gynecologists (ACOG) still endorses this terminology. Due in part to the increasing prevalence of obesity among young women, an increasing proportion of women will have unrecognized type 2 diabetes at the time of screening for gestational diabetes. To address this increased potential for prepregnancy diabetes, in 2010 the International Association of Diabetes and Pregnancy Study Group (IADPSG) recommended changing the classification of diabetes diagnosed during pregnancy to overt or gestational. The ADA and the World Health Organization (WHO) endorsed this recommendation. “Overt diabetes” (IADPSG) or “diabetes mellitus in pregnancy” (WHO) would be diagnosed at the initial prenatal visit (as long as the visit occurs in the first trimester) in women who met any of the following criteria: (1) fasting plasma glucose 126 mg/dL (7.0 mmol/L) or higher, or (2) 2-hour plasma glucose 200 mg/dL (11.1 mmol/L) or greater during an OGTT, or (3) hemoglobin A1C 6.5% (48 mmol/mol) or higher, or (4) random plasma glucose 200 mg/dL (11.1 mmol/L) or more in a patient with classic symptoms of hyperglycemia. Per the ADA, GDM would then be defined as “diabetes diagnosed in the second or third trimester of pregnancy that is not clearly either type 1 or type 2 diabetes.” ACOG does not address whether there should be a unique designation for women who have a positive diabetes screen in the first trimester of pregnancy.
Type 1 Diabetes Mellitus
Caused by β-cell dysfunction, usually leading to absolute insulin deficiency
Type 2 Diabetes Mellitus
Due to progressive loss of insulin secretion imposed on the background of insulin resistance
Gestational Diabetes Mellitus
Diabetes diagnosed in the second or third trimester of pregnancy that is not clearly overt diabetes
Other Types
Genetic defects in carbohydrate metabolism (i.e., maturity-onset diabetes of the young [MODY])
Diseases of the exocrine pancreas
Endocrinopathies
Drug- or chemical-induced diabetes
Infections
Uncommon forms of immune-mediated diabetes
Genetic syndromes sometimes associated with diabetes
Screening for Gestational Diabetes
Patients with GDM are typically asymptomatic. Many experts and organizations, including ACOG, the ADA, the WHO, and the US Preventive Services Task Force (USPSTF), among others, have recommended screening all pregnant women for GDM. The WHO has recommended using a 75-g, 2-hour OGTT for screening and diagnosis. The United Kingdom has also adopted the 2-hour OGTT for screening and diagnosis, but does not recommend universal screening. ACOG recommends universal screening, with a selective, risk-based approach to screening in the first trimester. In keeping with the 2013 Eunice Kennedy Shriver National Institute of Child Health and Human Development Consensus Development Conference on diagnosing gestational diabetes, ACOG recommends screening with a 50-g 1-hour GCT, followed by a 100-g 3-hour OGTT for diagnosis if GCT is positive. The most recent IADPSG study group recommendations suggest universal screening at 24 to 28 weeks’ gestation with the 75-g 2-hour OGTT, and screening high-risk women for pregestational diabetes with fasting or random plasma glucose, or hemoglobin A1C at the first prenatal visit. The low-risk category that some recommend excluding from GDM screening includes women under age 25 who have normal body mass index (BMI), who have no first-degree relatives with the condition, and who are not members of ethnic or racial groups with a high prevalence of diabetes (Hispanic, Native American, Asian, or African American).
Screening traditionally has been performed at 24 to 28 weeks of gestation. For women at risk for undiagnosed type 2 diabetes (women with a history of gestational diabetes or polycystic ovarian syndrome [PCOS], a high BMI, persistent glycosuria, a strong family history of diabetes, a prior macrosomic infant, or a prior unexplained late fetal demise), early screening for diabetes mellitus in pregnancy (or GDM, depending on which governing body’s nomenclature is being followed) should be performed at the first prenatal visit. If the early screen is negative, the screen should be repeated at 24 to 28 weeks.
Two-Step Approach
At this time, there is no universal accepted standard for screening and diagnosis of diabetes in pregnancy. Screening for diabetes in pregnancy has traditionally been performed as a two-step approach. The first step uses the glucose load test (GLT), also known as the glucose challenge test (GCT). First proposed as a screening test for GDM by O’Sullivan et al. in 1973, the GLT is a nonfasting 50-g oral glucose challenge followed by a venous plasma glucose measurement at 1 hour. The GLT is considered positive if the 1-hour glucose measurement is greater than a previously agreed threshold. Threshold values have been suggested to be 130, 135, and 140 mg/dL. Use of a lower cutoff will increase the detection rate of women with GDM, but will result in a substantial increase in the false-positive rate ( Table 27.2 ). There is no absolute GLT cutoff that should be regarded as diagnostic of GDM.
| Glucose Cutoff | Proportion of | Sensitivity for |
|---|---|---|
| Women With a | Gestational | |
| Positive Test | Diabetes Mellitus | |
| ≥140 mg/dL | ||
| (≥7.8 mmol/L) | 14%–18% | ~80% |
| ≥130 mg/dL | ||
| (≥7.2 mmol/L) | 20%–25% | ~90% |
The second step of the two-step approach requires a 3-hour glucose tolerance test (GTT), which is only performed if the GLT is positive. A fasting glucose measurement of at least 105 mg/dL in the setting of a positive GLT is highly predictive of an abnormal GTT. In pregnancy, the GTT involves an overnight fast and subsequent 100-g oral glucose challenge. Venous plasma glucose is measured fasting and at 1 hour, 2 hours, and 3 hours after the glucose load.
Although there is general agreement that two or more abnormal values are required to confirm the diagnosis, there is little consensus about the glucose values that define the upper range of normal in pregnancy and no threshold that perfectly predicts adverse pregnancy outcome. Even one elevated value has been associated with an increased incidence of macrosomia and birth injury.
A secondary analysis of a treatment trial for mild gestational diabetes found that elevations in fasting glucose and 3-hour GTT levels were associated with adverse pregnancy and neonatal outcomes. The same study found that fasting glucose of 90 mg/dL or greater and 1-hour value of 165 mg/dL or greater were associated with an increased risk for adverse neonatal outcomes, while a 1-hour value of 150 mg/dL or greater was associated with an increased risk for large for gestational age neonates.
Current diagnostic cutoffs for the 3-hour GTT are depicted in Table 27.3 .
| Plasma Glucose Values mg/dL (mmol/L) | |||
|---|---|---|---|
| National Diabetes | Carpenter and | ||
| Data Group * | Sacks et al. † | Coustan ‡ | |
| Fasting | 105 (5.8) | 96 (5.3) | 95 (5.2) |
| 1-h | 190 (10.6) | 172 (9.4) | 180 (9.9) |
| 2-h | 165 (9.2) | 152 (8.3) | 155 (8.6) |
| 3-h | 145 (8.1) | 131 (7.2) | 140 (7.7) |
* National Diabetes Data Group: Classification and diagnosis of diabetes and other categories of glucose intolerance. Diabetes 28:1039, 1979.
† Sacks DA, Abu-Fadil S, Greenspoon JS, Fotheringham N: Do the current standards for glucose tolerance testing in pregnancy represent a valid conversion of O’Sullivan’s original criteria? Am J Obstet Gynecol 161:638, 1989.
‡ Carpenter MW, Coustan DR: Criteria for screening tests for gestational diabetes. Am J Obstet Gynecol 144:768, 1982.
One-Step Approach
In 2010, the one-step approach to diagnosing diabetes in pregnancy was proposed by the IADPSG. This approach was subsequently endorsed by the ADA, the WHO, and the National Institute for Health and Care Excellence (NICE) in the United Kingdom, but not by ACOG. The IADPSG approach advocates diagnosing gestational diabetes in women who meet the criteria outlined in Box 27.2 : (1) fasting plasma glucose greater than or equal to 92 mg/dL (5.1 mmol/L) but less than 126 mg/dL (7.0 mmol/L) at any gestational age, or (2) at least one abnormal result on a 75 g 2-hour OGTT, which could be administered at 24 to 28 weeks of gestation. Abnormal values are defined as (1) fasting plasma glucose greater than or equal to 92 mg/dL (5.1 mmol/L) but less than 126 mg/dL (7.0 mmol/L); or (2) 1-hour value 180 mg/dL (10.0 mmol/L) or more; or (3) 2-hour value 153 mg/dL (8.5 mmol/L) or more (see Box 27.2 ). The IADPSG-selected thresholds for the 2-hour OGTT are based on data from the Hyperglycemia and Adverse Pregnancy Outcomes (HAPO) study, a multinational, multicenter prospective observational study of more than 23,000 pregnancies that demonstrated a continuous association between maternal glycemic concentrations and adverse perinatal outcomes. As maternal fasting plasma glucose levels increased from 75 mg/dL, and as the 1- and 2-hour oral GTT values increased, the risk of large-for-gestational-age infants, elevated cord blood C-peptide, neonatal hypoglycemia, and cesarean delivery increased continuously. The IADPSG-selected thresholds thus represent the average glucose values in the HAPO study, at which there were 1.75 times the odds of infant birth weight, cord blood C-peptide (a proxy for fetal hyperinsulinemia), and percent neonatal body fat greater than the 90th percentile. Women with one or more values above these thresholds had a twofold higher frequency of preeclampsia and large for gestational age infants, and more than a 45% increase in preterm delivery and primary cesarean delivery. The algorithm for screening and diagnosis of GDM proposed by IADPSG is outlined in Box 27.2 .
* Guidelines are to be applied to women without known diabetes antedating pregnancy. Postpartum glucose testing should be performed for all women diagnosed with overt diabetes during pregnancy or GDM.
First Prenatal Visit
- •
Measure FPG, A1C, or random plasma glucose on all or only high-risk women †
† Decision to perform evaluation for glycemia on all pregnant women at first prenatal visit or only on women with characteristics indicating a high risk for diabetes is to be made on the basis of the background frequency of abnormal glucose metabolism in the population and on local circumstances.
- •
If results indicate overt diabetes (FPG ≥ 7.0 mmol/L [126 mg/dL], A1C ≥ 6.5%, random plasma glucose ≥ 11.1 mmol/L [200 mg/dL] with confirmation):
- •
Treat and follow-up as for preexisting diabetes
- •
- •
If results not diagnostic of overt diabetes and FPG ≥ 5.1 mmol/L (92 mg/dL) but < 7.0 mmol/L (126 mg/dL):
- •
Diagnose as GDM
- •
- •
If results not diagnostic of overt diabetes and FPG < 5.1 mmol/L (92 mg/dL):
- •
Test for GDM from 24 to 28 weeks’ gestation with a 75-g OGTT ‡
‡ The IADPSG panel concluded there is insufficient evidence to know whether there is a benefit of generalized testing to diagnose and treat GDM before the usual 24 to 28 weeks’ gestation.
- •
- •
24–28 Weeks’ Gestation: Diagnosis of GDM
- •
2-h 75-g OGTT: perform after overnight fast on all women not previously found to have overt diabetes or GDM during testing earlier in this pregnancy:
- •
Diagnose overt diabetes if FPG ≥ 7.0 mmol/L (126 mg/dL)
- •
Diagnose GDM if one or more values equals or exceeds the following thresholds:
FPG ≥ 5.1 mmol/L or 92 mg/dL
1-h plasma glucose ≥ 10.0 mmol/L or 180 mg/dL
2-h plasma glucose ≥ 8.5 mmol/L or 153 mg/dL
- •
OGTT is normal if all values are less than above thresholds
- •
FPG , Fasting plasma glucose; GDM , gestational diabetes mellitus; IADPSG , International Association of Diabetes and Pregnancy Study Group; OGTT , oral glucose tolerance test.
The use of IADPSG diagnostic criteria for overt and gestational diabetes would result in 18% of women being diagnosed with diabetes in pregnancy. The potential long-term economic impact of implementing the IADPSG guidelines is unknown at this time, but in the short term, health care costs would likely be increased. Including the costs of long-term health intervention with diet and exercise, a cost-effectiveness analysis estimated that for every 100,000 pregnancies, the IADPSG approach would increase costs by more than $125,600,000. This analysis concluded that the IADPSG screening recommendations are cost-effective only if postdelivery care reduces diabetes incidence.
Adverse Effects of Maternal Hyperglycemia
Identification of overt diabetes early in pregnancy is important due to the associated increased risk of congenital anomalies, and maternal complications such as nephropathy and retinopathy. In contrast, GDM poses little immediate risk to the mother. Women with GDM are not at risk of diabetic ketoacidosis (DKA), which is primarily a disease of absolute insulin deficiency, but are at increased risk for developing hypertensive disorders of pregnancy. GDM has been associated with a variety of perinatal and neonatal complications, including increased risk of macrosomia, operative delivery, shoulder dystocia, birth trauma, hypoglycemia, hyperbilirubinemia, hypocalcemia, and perinatal mortality.
Transplacental glucose transport is a facilitated process that is mediated by the glucose transporter isoform GLUT-1. Some have hypothesized that maternal hyperglycemia in diabetes increases placental glucose transfer, resulting in fetal hyperglycemia and increased fetal insulin concentration that in turn stimulates fetal growth. However, even in diabetic pregnancies with evidence of strict glycemic control, fetal macrosomia (defined as an estimated fetal weight of at least 4500 g) is not uncommon, which suggests a complex relationship between metabolic derangement and fetal growth in diabetes. Other factors that may contribute to fetal macrosomia include obesity and high circulating levels of amino acids and lipids.
Many of the complications of GDM are due to fetal macrosomia. Increased birth weight is associated with an increased risk of cesarean delivery, operative vaginal delivery, and birth injury to both the mother (vaginal, perineal, and rectal trauma) and fetus (including orthopedic and neurologic injury). Shoulder dystocia with resultant brachial plexus injury is a serious consequence of fetal macrosomia, and risk is further increased in the setting of GDM because the macrosomia of diabetes is associated with increased diameters in the upper thorax of the fetus.
Recent data also suggest that the offspring of mothers with both gestational and prepregnancy diabetes may be predisposed to develop obesity, diabetes, and hypertension, via intrauterine programming/epigenetic modification. For example, a study of 9439 children aged 5 to 7 evaluated the prevalence of childhood obesity relative to maternal glycemia in pregnancy. This study found that maternal fasting glucose level of 95 mg/dL or more on a 3-hour 100-g OGTT, gestational diabetes by Carpenter and Coustan criteria, and a top quartile 50-g 1-hour GCT result were all associated with childhood obesity. Treatment of GDM reduced the rates of childhood obesity to rates similar to that of offspring born to women with normal GCTs, but this effect was only observed among offspring with birthweight of 4000 g or less. The authors concluded that maternal hyperglycemia resulted in metabolic imprinting for childhood obesity in offspring.
Interventions for GDM that definitively decrease the risk of macrosomia and subsequent adverse outcomes are limited. Two multicenter randomized clinical trials have demonstrated that the treatment of mild hyperglycemia in pregnancy reduces neonatal morbidity and macrosomia. The Australian Carbohydrate Intolerance Study in Pregnant Women Trial (ACHOIS) demonstrated that treating pregnant women with impaired glucose tolerance on a 75-g OGTT resulted in a significant reduction in a composite perinatal outcome, including perinatal death, shoulder dystocia, orthopedic injury, and nerve palsy. In the ACHOIS trial, glycemic control was achieved through a combination of dietary counseling, four times daily home blood glucose monitoring (maintaining fasting glucose levels <99 mg/dL and 2-hour postprandial levels <126 mg/dL), and insulin for persistent hyperglycemia. There was a reduction in the diagnosis of macrosomia (21% to 10%), but an increase in neonatal ICU admissions (61% to 71%). Induction of labor was increased in the intervention groups (from 29% to 39%), with no increase in the cesarean delivery rate, which was stable at 31% to 32%. In 2009, a second randomized controlled trial also demonstrated improved maternal and neonatal outcomes in women with glucose intolerance on a 100-g OGTT. Landon and colleagues reported that glycemic control achieved through a combination of dietary counseling, four times daily home blood glucose monitoring (maintaining fasting glucose levels <95 mg/dL and 2-hour postprandial levels <120 mg/dL), and insulin for persistent hyperglycemia did not result in a significant reduction in combined neonatal morbidity and mortality—a composite outcome including stillbirth, perinatal death, neonatal hyperbilirubinemia, hypoglycemia, hyperinsulinemia, and birth trauma. Treatment of glucose intolerance did, however, result in significant reductions in mean neonatal birth weight and fat mass, reduced frequency of large for gestational age neonates (14.5% to 7%), birth weight greater than 4000 g (14% to 6%), shoulder dystocia (4% to 1.5%), cesarean delivery (34% to 27%), and pregnancy-induced hypertension (14% to 9%), compared with usual care. A 2013 systematic review and meta-analysis found that treatment of GDM with dietary interventions or insulin administration (if blood glucose targets are not achieved with diet alone) resulted in significant reductions in preeclampsia (RR 0.62 [0.42 to 0.89]), shoulder dystocia (RR 0.42 [0.23 to 0.77]), and neonatal birth weight greater than 4000 g (RR 0.50 [0.35 to 0.71]). The only potential harm recognized from treatment of GDM in this meta-analysis was an increased number of prenatal visits.
Management of Gestational Diabetes Mellitus
The goal of antepartum management is to prevent fetal macrosomia and its resultant complications by maintaining maternal blood glucose at desirable levels throughout gestation (fasting, below 95 mg/dL; 1 hour postprandial, below 140 mg/dL; or 2 hours postprandial, below 120 mg/dL). Initial recommendations should include a diabetic diet consisting of 30 to 35 kcal/kg of ideal body weight, given as 40% to 50% carbohydrate, 20% protein, and 30% to 40% fat, to avoid protein catabolism. Daily home glucose monitoring and weekly antepartum visits to monitor glycemic control should also be instituted. If diet alone does not maintain blood glucose at desirable levels, insulin administration may be required. If initial fasting glucose levels are consistently greater than 95 mg/dL and/or postprandial values are consistently above 140 mg/dL (1 hour after starting a meal) or 120 mg/dL (2 hours after starting a meal), insulin therapy can be started immediately, with every effort made to avoid iatrogenic hypoglycemia. The Fifth International Workshop on Gestational Diabetes recommended exercise as an adjunct to diet for the treatment of GDM, although data are conflicting on the beneficial effects of exercise on glycemic control. Some randomized trials have failed to demonstrate a beneficial effect of exercise on glycemic control in women with GDM, but other studies have demonstrated that exercise is associated with a reduction in need for insulin in women with GDM, and a reduction in macrosomia and cesarean delivery rates. The ADA recommends including moderate exercise in the treatment of women with GDM and no medical or obstetric contraindications to physical activity.
ACOG and the ADA have recently endorsed the use of oral antihyperglycemic agents in pregnancy, although the US FDA has not specifically approved these medications for treatment of GDM. The ADA specifically recommends the use of insulin or metformin over glyburide, due to the higher rates of neonatal hypoglycemia and macrosomia reported with glyburide (discussed later).
In the United States, oral hypoglycemic agents (sulfonylureas in particular) have not traditionally been recommended as first-line agents for use during pregnancy because of the possibility of fetal teratogenesis and prolonged neonatal hypoglycemia. This class of drugs works by stimulating pancreatic β-cells to synthesize and release insulin. Because the adverse fetal consequences of GDM are likely related to fetal hyperinsulinemia, any agent that could cross the placenta and increase fetal insulin production should be used with caution in pregnancy. First-generation sulfonylureas have been shown to cross the placenta and, as such, are contraindicated in pregnancy. There are conflicting data regarding the transplacental passage of second-generation sulfonylurea agents (glyburide and glipizide). Older human studies demonstrated minimal fetal exposure, likely secondary to high protein binding and active transport of glyburide from the fetal to the maternal circulation. One study reported umbilical cord blood concentrations of glyburide as high as 70% of maternal serum concentrations, and another found significant variability in transplacental transfer of glyburide among patients, with 37% of cord blood samples containing higher glyburide concentrations than maternal blood. No data have been published on long-term effects of maternal glyburide use on offspring, and patients prescribed glyburide for treatment of GDM should be informed of uncertainties with respect to the extent of transplacental passage and long-term effects on offspring.
Congenital malformations associated with the use of oral sulfonylurea drugs in pregnancy have been described, but most of these reports failed to take into account maternal glycemic control. More recent studies have shown that the risk of malformations correlates strongly with the degree of glycemic control at the time of conception and is unrelated to the type of antidiabetic therapy. Moreover, such reports refer to oral hypoglycemic treatment in early pregnancy, whereas treatment for GDM only begins after the period of fetal organogenesis, thereby eliminating any concern regarding malformations due to treatment alone.
Between 1974 and 1983, Coetzee and Jackson treated 423 women with a new diagnosis of diabetes in pregnancy with oral hypoglycemic agents, and found no cases of serious neonatal hypoglycemia and no increase in perinatal mortality. A clinical trial by Langer et al. randomized 404 women with singleton pregnancies and GDM, requiring treatment to either glyburide or insulin therapy. Results showed no difference in glycemic control or neonatal outcome (including congenital malformations, macrosomia, neonatal hypoglycemia, or admission to neonatal intensive care). Eight women (4%) in the glyburide group required insulin therapy. Jacobson et al. performed a retrospective analysis comparing 236 women taking glyburide to 268 women taking insulin for control of gestational diabetes unresponsive to diet therapy. Women in the insulin group had a higher mean BMI and higher mean fasting value on GTT, suggesting these women may have had a greater predisposition to poor glycemic control. The study reported no significant differences between groups in birth weight, macrosomia, or cesarean delivery. More women in the glyburide group achieved mean fasting and postprandial goals (86% vs. 63%), and their neonates were less likely to be admitted to the NICU compared with women taking insulin (15% vs. 24%). Women treated with glyburide, however, had a higher incidence of preeclampsia (12% vs. 6%), and their neonates were more likely to receive phototherapy (9% vs. 5%). From the standpoint of maternal safety, hypoglycemia is the most commonly reported maternal side effect. Langer et al. reported that 2% of patients taking glyburide had blood glucose measurements less than 40 mg/dL, compared with 20% of patients taking insulin. Other studies reported no significant difference in rates of maternal hypoglycemia between women taking glyburide compared to insulin.
A 2015 systematic review and meta-analysis of randomized trials comparing glyburide to insulin for treatment of GDM found that women assigned to glyburide had a higher risk of macrosomia (RR 2.62 [1.35 to 5.08]), higher mean birthweight in offspring (mean increase of 109 g [36 to 181 g]), and a higher rate of neonatal hypoglycemia (RR 2.04, [1.3 to 3.2]). While the limited data currently available do not permit firm conclusions to be drawn about the efficacy and safety of oral hypoglycemic drugs in pregnancy, these agents are being used more frequently by obstetric care providers.
The largest randomized clinical trial to date investigating the efficacy of metformin compared with insulin for treatment of gestational diabetes is the Metformin in Gestational Diabetes Trial (MiG). In this randomized, open-label trial, 363 women were randomized to metformin and 370 to insulin, with 46% of women on metformin requiring supplemental insulin. There was no significant difference in perinatal complications between treatment groups. The primary composite outcome of neonatal hypoglycemia, respiratory distress, need for phototherapy, birth trauma, 5-minute Apgar score less than 7, or prematurity was 32% in both the metformin and the insulin group. Women preferred metformin to insulin therapy (77% vs. 27%), and there were no serious adverse events associated with the use of metformin.
A smaller open-label study randomized 100 women with GDM to therapy with insulin or metformin, finding no significant differences in the incidence of large for gestational age, mean birthweight, or neonatal morbidity. Thirty-two percent of women randomized to metformin required supplemental insulin; these women tended to have higher BMIs, higher fasting blood glucose levels, and required medical therapy for GDM earlier than those women who achieved adequate glycemic control with metformin alone.
The 2015 systematic review and meta-analysis comparing glyburide, insulin, and metformin for the treatment of GDM found that compared with use of insulin, metformin resulted in less gestational weight gain, but lower gestational age at delivery and higher risk of preterm birth. There were no statistically significant differences in mean birth weight or macrosomia between metformin and insulin users, but there was a nearly significant trend toward lower neonatal hypoglycemia in metformin users (pooled risk ratio 0.78 [0.6 to 1.01]). Thus metformin may provide a reasonable alternative to insulin in the treatment of gestational diabetes, particularly in lean or moderately overweight women who develop GDM later in gestation.
The use of metformin has several advantages over glyburide, including less macrosomia (RR 0.33 [0.13 to 0.81]), lower mean birth weight (mean difference −209 g, [−314 to −104 g]), and lower gestational weight gain (mean difference −2.06 kg [−3.98 to −0.14 kg]). Compared with women taking glyburide (4% to 16%), women using metformin are more likely (35% to 50%) to require supplemental insulin to achieve adequate glycemic control.
Metformin is known to cross the human placenta, which has led to some trepidation regarding its safety profile in pregnancy. Metformin is a biguanide agent that acts by reducing peripheral insulin resistance and inhibiting gluconeogenesis. Since insulin acts as a potent growth factor, there is a theoretical concern that metformin passage across the placenta may lead to excessive fetal growth. Despite these concerns, metformin is commonly used by reproductive endocrinologists to help achieve pregnancy in women with PCOS. In vitro models and in vivo studies have shown that metformin crosses from the maternal to the fetal compartment. In one study, umbilical cord blood levels of metformin were twice as high as maternal venous levels.
There are very few data on whether fetal exposure to an insulin-sensitizing agent like metformin is beneficial or harmful. Multiple small retrospective studies have failed to show any significant adverse outcome with first trimester metformin exposure. A follow-up study of offspring of the Metformin in Gestational diabetes trial (MiG TOFU) reported no difference between metformin-exposed and nonexposed offspring in neurodevelopmental outcomes, total fat, or central adiposity at 2 years, but did find an increase in subcutaneous fat deposition. Experts disagree on whether this may represent a healthier fat distribution in exposed offspring (i.e., if offspring exposed to metformin develop less visceral fat, they would theoretically be more insulin-sensitive), and longer-term studies are needed. Until longer-term follow-up data are available regarding the impact on offspring metabolic profile and neurodevelopment after in utero exposure to metformin, patients should be counseled regarding the uncertainty about the effects of transplacental passage.
The risk of stillbirth is increased in women with poorly controlled GDM, although it is not clear whether this is true also of pregnancies with mild disease. For this reason, many centers recommend weekly fetal testing starting at 32 weeks and early delivery (typically at 38 to 40 weeks) for women with GDM who require oral or insulin therapy and those with a pregnancy complication (macrosomia, polyhydramnios, or hypertension). Whether weekly fetal testing and early delivery is necessary in pregnancies complicated only by diet-controlled GDM is not clear. Sonographic estimation of fetal weight should be considered at 36 to 38 weeks’ gestation. The use of prophylactic elective cesarean delivery to reduce the risk of maternal and fetal birth injury in the setting of fetal macrosomia remains controversial. It is clear, however, that induction of labor for so-called impending macrosomia does not decrease the risk of cesarean delivery or intrapartum complications. In labor, maternal glucose levels in pregnancies complicated by GDM should be maintained at 100 to 120 mg/dL to minimize the risk of fetal hypoxic injury. For the same reason, neonatal blood glucose levels should be measured within 1 hour of birth, and early feeding should be encouraged. Delivery of the fetus and placenta effectively removes the source of the antiinsulin hormones that cause GDM. As such, no further management is required in the immediate postpartum period.
GDM frequently indicates underlying insulin resistance. Fifty percent of women with GDM will experience GDM in subsequent pregnancies, and 30% to 65% will develop type 2 diabetes later in life. All women with GDM should therefore have a standard (nonpregnant) 75-g GTT approximately 6 weeks postpartum and should consider preventative and early diagnostic strategies like weight reduction, increased exercise, and regular screening for diabetes.
Pregnancy in Women With Type 1 or Type 2 Diabetes Mellitus
- ◆
In contrast to gestational diabetes, pregestational diabetes is associated with significant maternal and perinatal morbidity and mortality.
- ◆
There is a significantly increased risk of congenital anomalies, particularly cardiac defects, neural tube defects, and renal agenesis. Fetal echo is indicated.
- ◆
Insulin, rather than oral agents, has been the mainstay of therapy in pregestational diabetics.
Pregestational diabetes, which affects approximately 1% of women of childbearing age, can be due either to absolute insulin deficiency (type 1, insulin-dependent diabetes mellitus [IDDM]) or to increased peripheral resistance to insulin action (type 2, non-insulin-dependent diabetes mellitus [NIDDM]), as shown in Box 27.1 . The fasting glucose cutoff for diagnosing pregestational diabetes was reduced from 140 to 126 mg/dL in 1997. On the basis of this change, there are more than 29 million people with diabetes in the United States alone. The White classification of diabetes in pregnancy ( Table 27.4 ) was developed by Dr. Priscilla White at the Joslin Diabetic Center in Boston, Massachusetts, in an attempt to correlate severity of diabetes with pregnancy outcome. Although this classification is commonly used, any direct correlation between White class and prognosis remains unclear. Features known to be associated with poor pregnancy outcome include DKA, poor compliance, hypertension, pyelonephritis, and vasculopathy.
| White Class | Age of Onset (Year) | Duration (Years) | Vascular Disease | Therapy |
|---|---|---|---|---|
| A | Only in pregnancy | Only in pregnancy | No | A1—diet controlled |
| A2—insulin requiring | ||||
| B | >20 or | <10 | No | Insulin |
| C | 10–19 or | 10–19 | No | Insulin |
| D | <10 or | >20 | Benign retinopathy; hypertension | Insulin |
| F | Any | Any | Nephropathy | Insulin |
| R | Any | Any | Proliferative retinopathy | Insulin |
| H | Any | Any | Atherosclerotic heart disease | Insulin |
| T | Any | Any | Renal transplant | Insulin |
In contrast to GDM, pregestational diabetes is associated with significant maternal and perinatal mortality and morbidity ( Box 27.3 ). One of the more important complications is the increased risk of congenital malformations associated with hyperglycemia at the time of fertilization and embryo development. The incidence of congenital anomalies and spontaneous abortions in such patients correlates directly with the degree of glycemic control at conception, as measured by circulating maternal glycosylated hemoglobin levels. Overall, approximately 30% to 50% of the perinatal mortality in diabetic pregnancy are due to fetal malformations. Structural anomalies commonly seen in association with diabetes include cardiac defects (ventricular septal defects, transposition of the great vessels), renal agenesis, and neural tube defects (anencephaly, open spina bifida). Some congenital defects—specifically sacral agenesis and caudal regression syndrome—are up to 400 times more common in the offspring of women with diabetes than in women with normal glucose metabolism and, as such, are considered pathognomonic. The overall prevalence of these anomalies, however, is low.
Maternal
- •
Spontaneous abortion
- •
Diabetic ketoacidosis
- •
Hypertension
- •
Preeclampsia/eclampsia
- •
Preterm birth
- •
Cesarean delivery
- •
Severe perineal injury
- •
Infectious morbidity (chorioamnionitis, endometritis, wound infection)
Fetal
- •
Congenital anomalies
- •
Macrosomia
- •
Intrauterine growth restriction
- •
Late fetal demise
- •
Non-reassuring fetal testing (previously referred to as “fetal distress”)
Neonatal
- •
Birth trauma (e.g., hypoxic ischemic cerebral injury; skull, clavicular, and long bone fractures; shoulder dystocia and brachial plexus injury)
- •
Hypoglycemia
- •
Neonatal sepsis
- •
Delayed organ maturation (respiratory distress syndrome, hyperbilirubinemia)
The factors responsible for diabetic embryopathy are not well defined, but glucose and ketone bodies such as β-hydroxybutyrate have both been implicated. Oxidative stress and apoptosis dysregulation have also been identified as potential mechanisms for diabetic embryopathy in animal models. Several prospective randomized studies have shown that strict glycemic control around the time of conception is effective in reducing the risk of congenital malformations in women with established diabetes. In one trial, intensive preconception management of diabetic women with vascular disease reduced the malformation rate from 19% to 8.5%. Unfortunately, most women with diabetes do not seek care prior to conception. Maternal serum α-fetoprotein estimation at 15 to 20 weeks’ gestation and a detailed sonographic fetal anatomic survey (with or without a fetal echocardiogram) at 18 to 22 weeks can be useful in screening for fetal malformations.
Intensive antepartum management should be initiated as early as possible and continued throughout gestation, with a view to maintaining maternal blood glucose at desirable levels (fasting, below 95 mg/dL; premeal, below 100 mg/dL; 1 hour postprandial, below 140 mg/dL; or 2 hours postprandial, below 120 mg/dL; during the night, not below 60 mg/dL; with an overall goal of mean capillary glucose levels of 100 mg/dL, corresponding to a glycated A1C ≤6%). Although the frequency of self-glucose monitoring recommended in pregnancy varies by organization, glucose should be monitored a minimum of four times daily (fasting, and 1 or 2 hours postprandial), and could be monitored as many as nine times daily (fasting, premeal, 1 or 2 hours postprandial, before bedtime, and at 3 a.m. if nocturnal hypoglycemia is suspected). ACOG and the ADA provide recommendations for not only postprandial but premeal blood glucose levels. If compliance with frequent fingersticks is in question, postprandial blood glucose levels have been shown to correlate more closely with adverse pregnancy and neonatal outcomes than fasting or premeal blood glucose levels. Initial recommendations for management of pregestational diabetes include a strict diabetic diet, regular exercise, daily home glucose monitoring, insulin treatment, and weekly antepartum visits to monitor glycemic control. Such an approach has been shown to decrease perinatal mortality from a baseline of 20% to 30% to approximately 3% to 5%.
Insulin, rather than oral agents, has been the mainstay of therapy in pregestational diabetics. To date, there are no randomized clinical trials to establish efficacy of oral agents in the management of pregestational diabetes in pregnancy. Insulin should be administered subcutaneously at 0.7 to as high as 2.0 units/kg (present pregnancy weight, morbidly obese women may require doses as high as 1.5 to 2.0 units/kg) per day in divided doses. Traditional recommendations have been to administer two-thirds of the total daily dose in the morning (60% NPH, 40% regular/rapid acting) and one-third in the evening (50% NPH, 50% regular/rapid acting). Rapid-acting insulin, namely lispro or aspart, achieves better glycemic control with fewer hypoglycemic episodes compared with regular insulin. Lispro or aspart has the additional benefit of convenience, as these rapid-acting insulins can be administered immediately premeal, while regular insulin has to be administered 20 to 30 minutes before a meal. Alternative regimens may include dividing the total daily insulin requirement into 50% basal and 50% prandial, with either half or two-thirds of the basal administered as NPH in the morning, and the other half or one-third of basal insulin administered as NPH before bedtime. The remaining 50% of total daily insulin requirement may be divided in thirds and administered as lispro or aspart before meals. Insulin doses should be adjusted by approximately 10% to 20% up or down in response to the results of capillary blood glucose monitoring. Care should be taken to avoid iatrogenic hypoglycemia due to excessive insulin administration.
In women with pregestational diabetes, assessment of thyroid function is recommended (6% of diabetic women have co-existing thyroid disease), and baseline liver and renal function tests (including 24-hour urinary protein quantification and creatinine clearance determination) should be performed at the first prenatal visit. An ophthalmologic examination should also be carried out every trimester. Glycosylated hemoglobin levels should be determined at a minimum every trimester, and as frequently as every 4 to 8 weeks throughout gestation. At any one time, approximately 5% of maternal hemoglobin is glycosylated, known as hemoglobin Al (HbA1). HbAlc refers to the 80% to 85% of HbA1 that is irreversibly glycosylated and is therefore a more accurate measure of glycemic control. Because red blood cells have a life span of around 120 days, HbAlc measurements reflect the degree of glycemic control over the past 3 months.
Hypertension, prematurity, and late fetal demise are the most common complications of pregnancy in diabetic women. Approximately 30% of diabetic women will develop hypertension in the third trimester. Pregnancy-induced hypertension often results in labor induction and is a major contributor to premature delivery in diabetic women. Sustained maternal hyperglycemia results in fetal hyperglycemia that leads, in turn, to fetal hyperinsulinemia and increased oxygen demand. As such, fetuses of diabetic mothers are at increased risk of antepartum hypoxic ischemic cerebral injury and late fetal demise. Another possible mechanism for fetal hypoxia in diabetic pregnancy is maternal vasculopathy and hyperglycemia leading to uteroplacental perfusion. Given the increased risk for fetal growth restriction, hypoxia, and fetal demise, in addition to serial third trimester evaluation of fetal growth, weekly antepartum fetal testing (fetal cardiotocography) is usually recommended, starting at 32 weeks’ gestation. After 36 weeks, testing is usually performed twice weekly. If the fetal heart rate tracing is abnormal, further testing (either a biophysical profile or contraction stress test) is mandatory.
A major issue in the care of the pregnant diabetic women is the proper timing of delivery. No pregnant diabetic woman should be delivered after 40 weeks because of the increased risk of late fetal demise. If glycemic control is good and there is no evidence of maternal vascular disease, spontaneous labor at term should be awaited. Women with poorly controlled diabetes or with complications (known vascular disease, worsening/new onset hypertension, IUGR, oligohydramnios), on the other hand, should be delivered between 37 and 39 weeks. Delivery as early as 34 weeks may be considered on an individual basis in patients with poor glycemic control. Early delivery in pregnancies complicated by pregestational diabetes is associated with an increased risk of fetal respiratory distress syndrome, and consideration should be given to the validation of fetal lung maturity prior to elective induction. Fetal lung maturation is inhibited by insulin and testosterone, and enhanced by endogenous cortisol, thyroxine, PRL, and estradiol-17β. In infants of diabetic mothers, hyperinsulinemia and hyperandrogenemia are common findings and may contribute to the delay in lung maturation observed in diabetic pregnancies. The increased testosterone observed in male infants of diabetic mothers may be due to elevated concentrations of human chorionic gonadotropin (hCG), which stimulates testosterone synthesis in fetal Leydig cells.
As many as 25% of infants of diabetic mothers are macrosomic. If the estimated fetal weight is at least 4500 g, many authorities would recommend elective cesarean delivery at or beyond 39 weeks to minimize the risk of birth trauma, primarily shoulder dystocia and resultant brachial plexus injury. Elective cesarean delivery in women with pregestational diabetes should be scheduled early in the morning, and the patient’s morning prandial insulin dose should be withheld. The usual nighttime dose of intermediate-acting (NPH) and rapid-acting insulin should be maintained prior to morning cesarean delivery, but if the patient uses a long-acting insulin at night such as detemir or glargine, 50% of the usual dose should be given the night before delivery.
In early labor, if oral intake is permitted, even consumption of 50% of the typical daily calories consumed will usually be sufficient to meet metabolic demands. During active labor, metabolic demand increases. Intravenous glucose should therefore be administered (typically 5% dextrose in half-normal saline at a rate of 75 to 100 mL per hour) to all women with pregestational diabetes in active labor (or to those in latent labor when oral intake is prohibited), and blood glucose levels should be checked every 1 to 2 hours. Regular insulin should be given as needed either by intravenous infusion (starting at 0.5 to 1 units per hour) or subcutaneous injection to maintain maternal glucose levels at greater than 70 and less than 120 mg/dL (goal range 100 to 120 mg/dL). Strict maternal glycemic control in labor is critical to preventing fetal hyperglycemia and hyperinsulinemia, both of which increase fetal oxygen demand and thereby predispose to fetal cerebral hypoxic ischemic injury.
During the first 48 hours postpartum, women may have a “honeymoon period” during which their insulin requirement is decreased. Moreover, the need for strict glycemic control is reduced, and circulating glucose levels of 150 to 200 mg/dL can be comfortably tolerated during this period pending discharge from the hospital and regulation of glucose levels in the home environment. Once a woman is able to eat, she can return to her prepregnancy insulin regimen.
Obesity and Pregnancy
- ◆
Obesity is associated with increased risk of adverse obstetric outcomes, including miscarriage, congenital malformations, stillbirth, preeclampsia, gestational diabetes, cesarean delivery, and VTE, among others.
- ◆
Limiting gestational weight gain and prepregnancy weight loss (sometimes through bariatric surgery) may mitigate maternal and fetal risks such as diabetes, hypertensive disorders of pregnancy, and macrosomia. However, maternal bariatric surgery has also been associated with an increased risk for small-for-gestational-age neonates.
- ◆
The neuroendocrine milieu of maternal obesity may contribute to the risk for metabolic malprogramming of the fetus, and subsequent increased risk of obesity and metabolic syndrome in the offspring of obese women.
Obesity is one of the greatest public health challenges in the United States, and throughout the world. The prevalence of obesity has steadily risen since the 1980s. In the United States, 37% of reproductive age women are obese, representing a 70% rise in prepregnancy obesity over the last decade. Obesity prevalence has also increased in Europe among women of reproductive age, although overall rates are lower (20% to 25%).
The preferred method of weight assessment is the BMI. BMI is calculated as the body weight in kilograms divided by the square of the height in meters. Normal weight is defined as a BMI between 18.5 and 24.9 kg/m 2 . Overweight refers to a BMI between 25 and 29.9 kg/m 2 . Obesity is a BMI greater than or equal to 30 kg/m 2 . Obesity is further divided into Class I (30 to 34.9 kg/m 2 ), Class II (35 to 39.9 kg/m 2 ), and Class III (BMI > 40 kg/m 2 ), also known as “morbid obesity” or “extreme obesity.” The advantage of using BMI for defining obesity is that no adjustments need to be made for gender or height, and no tables are required for determining the normal range.
Obesity is a complex neuroendocrine and metabolic disorder that has been implicated in a large number of fetal and maternal complications, including spontaneous abortion, congenital malformations, stillbirth, preeclampsia, GDM, fetal macrosomia, cesarean delivery, venous thromboembolic disease, surgical complications, and urinary tract infections.
Congenital malformations classically associated with obesity include neural tube defects, ventral wall defects, and abnormalities of the great vessels. There is some evidence suggesting an increased risk for other anomalies in offspring of obese women, including hypospadias, isolated hydrocephalus, and orofacial clefts.
In one study, obese women weighing more than 110 kg had a fourfold increased risk of having a fetus with a neural tube defect, as compared with a control population weighing 50 to 59 kg. For women weighing 80 to 89 kg, the risk was increased 1.9-fold. Interestingly, folic acid supplementation (0.4 mg daily) did not appear to reduce the risk of neural tube defects in this cohort of obese women. These data are consistent with other studies showing that a BMI greater than 29 kg/m 2 is associated with a 1.9-fold increase in the risk for neural tube defects.
The mechanism by which obesity causes congenital anomalies is not known. It is conceivable that, in obese women, subtle abnormalities in glucose metabolism contribute to the increased risk of congenital malformations, similar to that of pregestational diabetes.
Perinatal mortality is increased with progressive obesity. In a cohort of 167,750 Swedish women, Cnattingius and colleagues demonstrated a 1.7-fold increased risk in late fetal death for overweight women (BMI 25 to 29.9 kg/m 2 ) and a 2.7-fold increase for obese women (BMI >30 kg/m 2 ) when compared with women with a prepregnancy BMI less than 20 kg/m 2 . Obesity has consistently been associated with hypertensive disorders of pregnancy.
For example, one large prospective multicenter cohort study of more than 20,000 women demonstrated that obese women with a BMI between 30 and 34.9 kg/m 2 had a 2.5- and 1.6-fold relative risk for gestational nonproteinuric hypertension and preeclampsia, respectively. For obese women with a BMI greater than 35 kg/m 2 , a similar association was found, with a relative risk of 3.0- and 3.3-fold, respectively.
In a systematic review of 13 studies including more than 1.4 million women, O’Brien et al. calculated a 2.0-fold increased risk of developing preeclampsia with every 5 to 7 kg/m 2 increase in BMI.
Other well-documented risks of obesity include an increased incidence of GDM and fetal macrosomia, birth injury, maternal perineal trauma, and cesarean delivery. In a study of 20,130 births, a BMI greater than 39 kg/m 2 was associated with a 46% cesarean delivery rate compared with 20% in a control group of women with a BMI less than 29 kg/m 2 . The increased cesarean delivery rate is also associated with increased surgical morbidity in obese women, including anesthetic complications, wound separation and infection, and venous thromboembolic events. Interestingly, maternal obesity appears to be relatively protective against spontaneous preterm birth. The risk of spontaneous preterm birth decreases with increasing maternal BMI, perhaps due to decreased uterine activity in these women compared with normal BMI controls.
Obstetric management of the obese patient should include calculation of BMI, careful attention to blood pressure, a nutrition consultation, institution of a daily exercise program, and early screening for GDM. Randomized controlled trials seeking to establish the optimum gestational weight gain in obese women are ongoing. Limiting total pregnancy weight gain to no more than 15 to 25 pounds appears to decrease the risk of fetal macrosomia without increasing the risk of low birth weight or IUGR. A careful sonographic anatomy survey (with or without fetal echocardiogram) at 18 to 22 weeks’ gestation is indicated in obese women, given the increased risk of fetal structural anomalies, although body habitus may result in suboptimal imaging. Serial growth scans should also be considered, given the limitations of other methods of fetal growth assessment. Anesthesia consultation in the third trimester should also be considered prior to the onset of labor. If cesarean section is required, every effort should be made to reduce the risk of wound separation and infection, including prophylactic antibiotics, and closure of the subcutaneous layer.
Obesity has become one of the leading endocrine causes of morbidity and mortality in women of reproductive age. Bariatric surgery, which includes a number of procedures to reduce gastric capacity or bypass the stomach, has been shown to promote weight loss and reduce long-term mortality in morbidly obese patients. Its use as a weight loss tool has increased in popularity, and the resultant weight loss often leads to improved fertility. The safety of pregnancy after these procedures is unclear. Early case reports suggested an increased risk of adverse pregnancy outcomes. However, more recent studies, including a meta-analysis, have suggested a number of potential benefits, including reduced risk of diabetes, hypertensive disorders of pregnancy, and macrosomia. A large population-based cohort study including 670 Swedish women who underwent bariatric surgery, with up to five matched control pregnancies for each case, reported that bariatric surgery prior to pregnancy was associated with reduced risk of gestational diabetes (OR, 0.25, 95% CI, 0.13 to 0.47) and large-for-gestational-age infants (OR, 0.33, 95% CI, 0.24 to 0.44), with no significant change in the risk of preterm birth. More concerning, however, was the higher risk of small-for-gestational age infants in the women who underwent surgery (OR, 2.2, 95% CI, 1.64 to 2.95) and the nearly significant increased risk for stillbirth or neonatal death in women who underwent surgery (OR, 2.39, 95% CI, 0.98 to 5.85). The link between prior bariatric surgery and possible increased neonatal mortality merits further study. Women who have undergone bariatric surgery are at risk for malabsorption and vitamin deficiencies, so levels should be followed during pregnancy, and all these patients should receive vitamin and mineral supplementation—especially iron, folate, and vitamin B 12 .
Leptin is a hormone secreted primarily by adipocytes and acts to suppress appetite while increasing energy expenditure, thereby regulating body weight.
Mice lacking the leptin gene ( ob/ob mice) are obese and anovulatory. Administration of human leptin to ob/ob mice increases energy expenditure, reduces weight and fat mass, and restores ovulation and fertility. In humans, absolute leptin deficiency is rare, and supplemental recombinant leptin has not been shown to promote weight loss.
During pregnancy, leptin is produced by maternal and fetal adipocytes as well as the syncytiotrophoblast. Circulating leptin levels rise rapidly in the first trimester, maintain high levels throughout pregnancy, and drop precipitously after delivery, suggesting a major contribution from the placenta. Leptin levels in the maternal circulation correlate with maternal body mass, but not with levels in umbilical cord blood at birth or birth weight. However, leptin levels in umbilical cord blood do correlate directly with both birth weight and fetal adiposity. The significance of the increased leptin levels in the maternal circulation during pregnancy are unclear, although it has been suggested that leptin may act to mobilize maternal fat stores and increase the availability of substrates to the fetus.
In diabetic pregnancies, alterations in maternal and fetal leptin levels have not been particularly informative. In pregnancies complicated by preeclampsia, umbilical cord leptin levels are decreased and reflect the reduction in fetal growth and fat stores. Interestingly, maternal leptin levels are increased in preeclampsia and appear to correlate with the severity of the disease. The significance of this observation remains unclear.
Although the regulation and mechanisms of action of leptin are not fully understood, it is possible that manipulation of the leptin-leptin receptor system may ultimately prove to be a safe and effective treatment for adult obesity and/or fetal macrosomia.
The developmental origins of health and disease hypothesis posit that the in utero environment induces fetal adaptive responses to facilitate offspring health and survival. The metabolic and hormonal changes that result from these responses lead to altered homeostatic set points, which ultimately may prove maladaptive in other environments. Fetal adaptation is likely mediated by epigenetic phenomena, including DNA methylation and histone modification. There is growing evidence that maternal obesity/overnutrition may induce fetal adaptive responses that predispose to obesity, diabetes, metabolic derangements, and increased cardiovascular risk later in life. Maternal hyperglycemia, an inflammatory intrauterine milieu, and leptin dysregulation all may contribute to the increased cardiometabolic risk for offspring born to obese gravidas.
Sheep and rat models suggest that in utero overnutrition may predispose to childhood and adult obesity via increased leptin exposure, with subsequent inhibited leptin receptor development in the hypothalamus, and thus decreased sensitivity to leptin-mediated appetite suppression. Indeed, increased leptin and insulin concentrations have been described in human offspring of obese mothers, suggesting that fetal programming of appetite and satiety pathways may be an important determinant of adult disease states.
Hypothalamic-Pituitary Diseases
- ◆
Pregnancy is associated with multiple physiological changes in the hypothalamic-pituitary axis, including increased pituitary weight and volume, increased proportion of lactotrophs in the adenohypophysis, increased circulating levels of PRL, increased ACTH, increased total and free cortisol, increased maternal serum GH, and possible mild thyroid glandular hypertrophy.
- ◆
Pituitary tumors are classified as either microadenomas (<10 mm in diameter) or macroadenomas (>10 mm).
- ◆
Microadenomas typically have a benign course in pregnancy, while macroadenomas are more often associated with extrasellar extension, local invasion, or compression of the optic chiasm.
- ◆
Prolactinoma is the most common pituitary tumor in pregnancy. First-line therapy is typically medical, and includes the dopamine agonists bromocriptine and cabergoline.
Pregnancy-Associated Changes in Pituitary Structure and Function
The pituitary gland is composed of three parts: the anterior lobe (adenohypophysis), the intermediate lobe (prominent in the fetus but attenuated in the adult), and a posterior lobe (neurohypophysis). During pregnancy, the structure and function of the pituitary gland are significantly altered. In the nonpregnant state, the pituitary gland weighs between 0.5 and 1.0 g. One autopsy study of 118 pregnant women demonstrated a 30% increase in the weight of the pituitary gland compared with nonpregnant controls (1070 mg vs. 820 mg, respectively). This increase in weight is associated also with an increase in volume ( Table 27.5 ) and a change in shape. In pregnancy, the pituitary gland develops a convex, dome-shaped superior surface, which may impinge on the optic chiasm and account, in part, for the bitemporal hemianopia observed in some apparently healthy pregnant women. Pregnancy is not associated with an increased incidence of pituitary adenoma.
| Gestational Age (Weeks) | Subject Number | Pituitary Volume (mm 3 ) Mean ± SEM |
|---|---|---|
| Nonpregnant | 20 | 300 ± 60 |
| 9 | 10 | 437 ± 90 |
| 21 | 11 | 534 ± 124 |
| 37 | 11 | 708 ± 123 |
On the basis of the hormones they produce, the adenohypophysis contains at least five different cell types: lactotropes (that primarily secrete PRL), corticotropes (ACTH), somatotropes (GH), gonadotropes (luteinizing hormone [LH] and follicle-stimulating hormone [FSH]), and thyrotropes (thyroid-stimulating hormone [TSH]). The cellular composition of the adenohypophysis, however, changes throughout pregnancy. This is especially true of the lactotrope cell population. Immunohistochemical studies have shown that, in the nonpregnant state, approximately 20% of cells in the adenohypophysis are lactotropes. This number increases in pregnancy such that, by the third trimester, approximately 60% are lactotropes. Moreover, the increase in lactotropes is most pronounced in the lateral portions of the adenohypophysis. By 1 month postpartum, the number of lactotropes in the anterior pituitary of nonlactating women is decreased. However, postpartum resolution of lactotrope hyperplasia is incomplete, and nonpregnant multiparas have on average more lactotrope cells than nulligravid women.
In contrast to lactotropes, the numbers of somatotropes, gonadotropes, and α-subunit–secreting cells in the adenohypophysis decrease in pregnancy, and the number of thyrotropes do not change. These changes in cellular composition are associated with changes in circulating hormone levels.
Prolactin
Levels of PRL in the maternal circulation increase throughout pregnancy, reaching concentrations of approximately 140 ng/mL at term ( Fig. 27.8 ). Although the maternal decidua is a major site of PRL production during pregnancy, PRL in the maternal circulation originates primarily from the maternal pituitary, with small contributions from the maternal decidua and fetal pituitary. The observation that circulating levels of PRL in pregnant women with preexisting hypopituitarism remain low throughout pregnancy supports the hypothesis that very little decidual PRL enters the maternal circulation. Decidual production of PRL leads to elevated levels in the amniotic fluid, which peaks at approximately 6000 ng/mL at the end of the second trimester.
The hyperprolactinemia of pregnancy is likely due to increased circulating levels of estradiol-17β. Aside from this increase in basal levels, PRL secretion by the maternal pituitary is stimulated by thyrotropin-releasing hormone (TRH), arginine, meals, and sleep in a manner similar to that seen in nonpregnant women. After delivery, maternal PRL concentrations in nonlactating women decrease to prepregnancy levels within 3 months. In lactating women, basal circulating PRL levels decrease slowly to nonpregnant levels over a period of several months with intermittent episodes of hyperprolactinemia in conjunction with nursing.
Pregnancy is also associated with a shift in PRL isoforms. In the nonpregnant state, the N-linked glycosylated isoform of PRL (G-PRL) predominates in the circulation. As pregnancy progresses, increasing amounts of nonglycosylated PRL appear in the circulation. In the third trimester, the concentration of circulating nonglycosylated PRL exceeds that of GPRL. Nonglycosylated PRL appears to be more biologically active than G-PRL. The precise function of the elevated circulating levels of PRL in pregnancy is not clear, but it appears to be important in preparing breast tissue for lactation by stimulating glandular epithelial cell mitosis and increasing production of lactose, lipids, and certain proteins. The role of PRL in amniotic fluid is not known.
Pregnancy produces changes in PRL secretion that persist long after delivery. Musey and colleagues reported that the basal serum PRL level and the PRL response to perphenazine stimulation was lower after pregnancy than before pregnancy. They also found that the serum PRL concentration was significantly lower in the parous women (mean, 4.8 ng/mL) than in the nulliparous women (8.9 ng/mL). These and other studies suggest that pregnancy permanently suppresses the secretion of PRL by the maternal pituitary.
Adrenocorticotropic Hormone
ACTH levels in the maternal circulation increase from approximately 10 pg/mL in the nonpregnant state to 50 pg/mL at term, and increase further to approximately 300 pg/mL in labor ( Fig. 27.9 ). Although the placenta can produce ACTH, the majority of circulating ACTH appears to come from the maternal pituitary. Placental production of CRH may be a major cause of the elevated levels of ACTH in the maternal circulation. In nonpregnant women, serum CRH levels range from approximately 10 to 100 pg/mL. In the third trimester of pregnancy, these concentrations increase to 500 to 3000 pg/mL but then decrease precipitously after delivery.
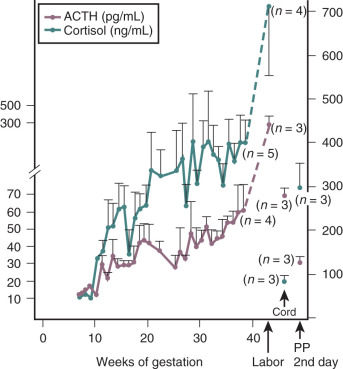
In addition to increasing pituitary ACTH secretion, chronically elevated levels of CRH in the maternal circulation reduce the ability of exogenous glucocorticoids to suppress the maternal ACTH–cortisol axis, enhance the ability of vasopressin to induce an ACTH response, and diminish the effect of exogenous CRH. CRH binding protein (CRH-BP) inactivates CRH, thereby preventing its action on the maternal or fetal pituitary. CRH-BP levels in the maternal circulation decrease during the last few weeks of pregnancy, resulting in an increase in free (biologically active) CRH.
Although the maternal adrenal glands do not change in size, pregnancy is associated with significant changes in the circulating concentrations of adrenal hormones. For example, serum cortisol levels increase substantially in pregnancy. The majority of circulating cortisol is bound to cortisol-binding globulin (CBG), which is produced by the liver. Circulating levels of CBG increase during pregnancy in response to elevated levels of estrogen, and CBG may retard the clearance of these hormones. It is not surprising, therefore, that total cortisol levels increase in pregnancy. However, levels of free cortisol in the circulation —as well as in saliva and urine —are also increased, which is likely due to the increased levels of ACTH in the maternal circulation. Maternal hypercortisolemia is also observed in complete molar pregnancy, suggesting that the increased cortisol is not derived from a fetal source. Other changes in adrenal hormone levels that occur in association with pregnancy include an increase in circulating levels of aldosterone and adrenal androgens, primarily androstenedione and testosterone.
Growth Hormone
Maternal serum GH levels begin to increase at around 10 weeks’ gestation, plateau at approximately 28 weeks, and can remain elevated for several months postpartum. The majority of this GH is derived from the placenta (GHV), with a marked reduction in basal somatotropin (GH-N) production by the maternal pituitary. Moreover, the release of GH-N in response to either insulin-induced hypoglycemia ( Fig. 27.10 ) or arginine stimulation is markedly attenuated, suggesting that maternal pituitary GH secretory reserve is diminished in pregnancy.
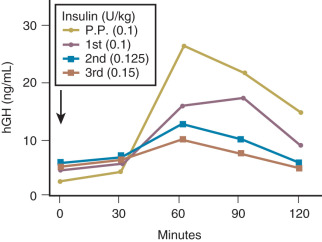
Thyroid-Stimulating Hormone
In general, the concentration of TSH (thyrotropin) in the maternal circulation remains within the normal range during pregnancy. Normative data from pregnant women suggest the upper reference range for TSH in pregnancy may be 2.5 to 3.0 mIU/L, rather than 4.0 mIU/L, the upper limit of normal in healthy, nonpregnant individuals. At 9 to 13 weeks of gestation, there is a modest decline in circulating TSH levels ( Fig. 27.11 ). This coincides with the peak placental production of hCG, and some authorities have suggested that the decrease in TSH may be due to the weak thyrotropic properties of hCG. An alternative hypothesis is that the placenta may secrete a hormone with TRH or TSH-like properties, but this hypothesis is not supported by most data.
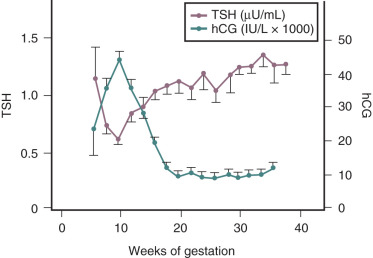
Enlargement of the thyroid gland is a common finding during pregnancy. This growth is not due to any deficiency in the hypothalamic-pituitary-thyroid axis, but rather a result of relative iodide deficiency and increased demand for thyroid hormone secondary to elevated levels of thyroid-binding globulin (TBG). Increased TBG concentrations result in the need for increased production of thyroxine (T 4 ) and triiodothyronine (T 3 ) to maintain adequate concentrations of free hormones. The thyroid responds to the need for increased production with increased vascularity, cellular hyperplasia, and ultimately glandular hypertrophy. Indeed, the TSH response to exogenous TRH stimulation remains normal throughout pregnancy. An appreciation of the physiologic changes in thyroid hormone levels is important to accurately assess thyroid status in pregnancy. Total T 4 and T 3 levels are elevated due to TBG excess, and therefore have not typically been used to evaluate thyroid status during pregnancy. The most appropriate test to detect thyroid dysfunction during pregnancy is the TSH assay. If this is abnormal, classically free T 4 and free T 3 levels have been measured. However, more recent uncertainty about the accuracy of free T 4 immunoassays in pregnancy has led some to reconsider the use of adjusted total T 4 in pregnancy in select situations (see the section on “ Maternal Thyroid Function in Pregnancy ”).
Gonadotropins
Maternal serum LH and FSH levels are decreased by 6 to 7 weeks of pregnancy and are below the limits of detection of many radioimmunoassays by the second trimester. The marked decrease in gonadotropin immunoreactivity in the pituitary glands of pregnant women coupled with the blunted LH and FSH response to exogenous gonadotropin-releasing hormone (GnRH) stimulation ( Fig. 27.12 ) suggest that this effect is localized primarily to the pituitary. The suppression of pituitary LH and FSH synthesis and secretion likely results from elevated circulating levels of sex steroids (estradiol-17β, progesterone) and regulatory peptides (such as inhibin) during pregnancy.
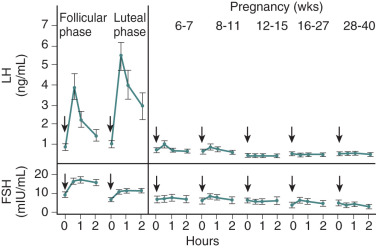
Pituitary Tumors in Pregnancy
Mutations are the primary cause of pituitary tumors. Most pituitary tumors are monoclonal, indicating that a somatic mutation in a single progenitor cell is the cause of the tumor. In one study, 100% of GH-producing tumors and 75% of ACTH-producing tumors were found to be monoclonal. In one series of GH-secreting tumors, mutations in the gene coding for the Gs protein were reported in 10 of 25 tumors, resulting in a constitutively active mutant Gs protein. Other endocrine factors (such as the levels of estradiol-17β, progesterone, and dopamine) can influence tumor phenotype, and changes in these hormones during pregnancy may affect tumor growth. In general, pituitary tumors are benign and slow-growing.
Pituitary tumors are commonly classified according to size as either microadenomas (<10 mm in diameter) or macroadenomas (larger than 10 mm). The clinical behavior of microadenomas and macroadenomas vary considerably during pregnancy. Macroadenomas may be associated with extrasellar extension, local invasion, or compression of the optic chiasm with resultant bitemporal hemianopia, and such conditions may become exacerbated in pregnancy. In one series of 60 pregnant women with macroadenomas, for example, 20% showed evidence of worsening visual field defects, significant enlargement on serial imaging studies, or neurological signs. Urgent neurosurgical decompression may be required during pregnancy if the tumor enlarges markedly or causes neurological sequelae.
In contrast, microadenomas tend to behave in a relatively benign manner in pregnancy, with no evidence of functional pituitary deficiency and a low risk of neurologic complications. In a longitudinal observational study of 215 pregnant women with microadenomas, for example, approximately 5% of women developed headaches, and less than 1% experienced worsening of visual field defects or demonstrated neurologic signs.
Prolactinoma
Prolactinoma refers to a tumor of PRL-secreting lactotrope cells, and is typically associated with elevated levels of PRL in the maternal circulation (see Chapter 3 ). In the initial evaluation of a suspected prolactinoma, measurement of circulating levels of thyroxine, TSH, and IGF-1 is important. This evaluation will exclude secondary causes of hyperprolactinemia, specifically hypothyroidism (thyroxine, TSH) and acromegaly (IGF-1). An imaging study of the hypothalamus and pituitary is also indicated, and computerized evaluation of the visual fields via automated perimetry is recommended if compression of the optic chiasm is suspected.
Women with marked hyperprolactinemia are usually anovulatory and, as such, infertile. If such a patient does not desire pregnancy, treatment with combination estrogen–progestin therapy will reduce the risk of osteoporosis and regulate the menstrual cycle. This approach appears to be safe and is associated with few tumor-related complications, including minimal risk of tumor growth. For infertile women with significant hyperprolactinemia who wish to conceive, treatment is usually required to induce ovulation. Controversy continues as to whether surgery or dopamine-agonist treatment represents the best first-line therapy for such women. Some authorities would recommend surgical treatment prior to conception to reduce both the need for dopamine-agonist treatment and the incidence of neurologic complications during pregnancy. However, microsurgical resection of a prolactinoma can result in death (in 0.3% of cases) or serious morbidity, such as a cerebrospinal fluid leak (0.4%). Moreover, a long-term cure can be expected in only approximately 60% of women treated surgically. For these reasons, the weight of evidence in the literature suggests that medical treatment should be regarded as the best first-line therapy for infertile women with significant hyperprolactinemia.
Having confirmed the diagnosis of a pituitary microprolactinoma, the goals of treatment are fourfold:
- 1.
Suppress PRL production and induce ovulation
- 2.
Decrease tumor size
- 3.
Preserve pituitary reserve
- 4.
Prevent tumor recurrence
Treatment with a dopamine agonist can normalize circulating PRL levels, establish regular ovulation, decrease tumor size, and preserve pituitary reserve. A disadvantage of dopamine-agonist treatment is that it is not effective in preventing tumor recurrence once treatment is discontinued. Four dopamine agonists have been demonstrated to be effective in the treatment of hyperprolactinemia: bromocriptine, pergolide, quinagolide, and cabergoline. Cabergoline is administered once weekly and may be more effective than bromocriptine in the treatment of microadenomas.
The two most commonly used dopamine agonists in pregnancy are cabergoline and bromocriptine. Information about the safety of cabergoline in pregnancy is more limited than information about bromocriptine, though data are available on more than 900 cases of periconceptional or first trimester cabergoline use, with no evidence of increased risk of spontaneous abortion, premature delivery, or multiple pregnancies. With respect to risk for congenital malformations with cabergoline, outcome data are available for 822 pregnancies, with a 2.4% rate of major malformations (not increased from baseline). There is more experience with the safety of bromocriptine in early pregnancy, with no data suggesting a significant increase in the rate of spontaneous abortions, multiple pregnancies, and fetal congenital abnormalities. The most common adverse effects associated with bromocriptine therapy are nausea, vomiting, and postural hypotension. Starting with low-dose therapy (0.625 mg daily) and increasing the dose slowly over a period of a few weeks can minimize these side effects. In some patients, doses as low as 2.5 mg daily may be effective. While bromocriptine has been more widely used in pregnancy, as experience with cabergoline accumulates, some experts prefer cabergoline over bromocriptine for treatment of prolactinoma in pregnancy, due to its better efficacy with fewer adverse effects. PRL levels should initially be checked every month for 3 months and thereafter every 3 months until the levels have returned to normal.
In women with microprolactinomas, bromocriptine or cabergoline can be discontinued once pregnancy is established. The majority of such women will have no further complications during pregnancy. For those women who do experience neurological sequelae such as headache or cranial nerve dysfunction, dopamine agonist treatment can be immediately reinstituted. Marked enlargement of a microprolactinoma or persistence of neurological sequelae despite medical treatment may be an indication for urgent neurosurgical intervention, but such complications are rare. In contrast, pituitary insufficiency and neurosurgical complications are far more common in women with macroadenomas. Such women should therefore be evaluated for panhypopituitarism before dopamine-agonist treatment is initiated. Women with macroprolactinomas are also more likely to develop complications in pregnancy. One approach to the management of such women is to discontinue dopamine agonists once pregnancy is established, and to reinstitute therapy if symptoms or signs of increasing tumor volume develop. An alternative plan is to continue dopamine agonist therapy throughout pregnancy. Lactation does not appear to worsen the clinical course of women with prolactinomas, and such women should be encouraged to breastfeed.
Cushing Disease
Cushing disease refers to the clinical syndrome resulting from excessive pituitary ACTH production. It is typically associated with depressed gonadotropin secretion, and spontaneous pregnancy is rare in women with untreated Cushing disease. Most cases of Cushing disease are due to pituitary microadenomas. As such, neurosurgical complications are rarely seen in pregnancy. However, the metabolic derangements associated with Cushing disease have been implicated as the cause of the observed increases in pregnancy-related complications, including premature labor, pregnancy-induced hypertension, and GDM.
Acromegaly
Acromegaly refers to the clinical syndrome associated with elevated circulating levels of GH. Acromegaly is often associated with anovulation, but spontaneous pregnancy can occur. Except for complications associated with pituitary enlargement, acromegaly does not appear to adversely affect pregnancy outcome. Since GH is an insulin antagonist, pregnancies complicated by excess circulating GH are at increased risk of hyperglycemia and diabetes. In most women, definitive treatment for acromegaly can be deferred until after delivery. Bromocriptine, transsphenoidal surgery, and more recently octreotide, a somatostatin agonist, have been successfully used to treat acromegaly during pregnancy.
Pituitary Insufficiency
Sheehan Syndrome
Sheehan syndrome (pituitary apoplexy) refers to the onset of acute hypothalamic–pituitary dysfunction that typically occurs after severe obstetric hemorrhage and resultant maternal hypotension at delivery. It is the most common cause of hypopituitarism worldwide, though not commonly seen in the United States. During pregnancy, the pituitary volume increases by approximately 100%. This increase in pituitary size, coupled with the low-flow, low-pressure nature of the portal circulation, appears to make the pituitary and parts of the hypothalamus particularly susceptible to ischemia caused by obstetric hemorrhage and hypotension. The majority of cases of Sheehan syndrome occur in developing countries where deliveries are not performed in health care facilities by skilled attendants, increasing the risk of complications from obstetric hemorrhage.
The hallmark of this syndrome is a loss of anterior pituitary hormone reserve, which may be complete or partial. PRL and GH deficiency are the most common abnormalities observed in Sheehan syndrome, but every imaginable pattern of pituitary hormone deficiency has been described. In a study of 10 African women with Sheehan syndrome, Jialal and co-workers described the pituitary hormone response to a combined intravenous insulin (0.1 unit/kg), TRH (200 mg), and GnRH (100 mg) challenge test. The pattern of pituitary hormone response revealed the following loss of secretory reserve: 100% of these women had both PRL and GH deficiency, 90% had cortisol deficiency, 80% had TSH deficiency, 70% had LH deficiency, and 40% had FSH deficiency.
The initial clinical manifestations of Sheehan syndrome include failure of lactation, failure of hair growth over areas shaved for delivery, poor wound healing after cesarean delivery, and generalized weakness. The best single test to confirm the diagnosis of Sheehan syndrome is to administer intravenous TRH (100 mg) and measure serum PRL levels at 0 and 30 minutes. The ratio of PRL measured at 30 minutes to that before TRH treatment (time 0) should be greater than 3.0. If the ratio is abnormal, a complete evaluation for panhypopituitarism should be initiated.
In addition to loss of anterior pituitary hormone reserve, mild hypothalamic and posterior pituitary dysfunction is also frequently seen in women with Sheehan syndrome. Detailed neuropathologic reports of autopsy specimens by Sheehan and Whitehead have shown that 90% of women with postpartum hypopituitarism have evidence of atrophy and scarring of the neurohypophysis. Subsequent studies have also demonstrated atrophy of the supraoptic and paraventricular nuclei in such patients. These observations have been confirmed in several clinical studies demonstrating that most women with Sheehan syndrome have mild functional defects in both vasopressin secretion and maximal urinary concentrating capability.
Lymphocytic Hypophysitis
Lymphocytic hypophysitis is a rare disorder caused by infiltration of the adenohypophysis with lymphocytes and plasma cells. Most cases of lymphocytic hypophysitis occur in women in the third trimester of pregnancy or immediately postpartum. In some cases, circulating antipituitary, antinuclear, or antimitochondrial antibodies have been detected. Pituitary enlargement can result in neurologic complications (headache, visual field defects, cranial nerve palsy) requiring surgical intervention, while pituitary cell damage may result in hyperprolactinemia, hypothyroidism, or adrenal insufficiency. High-dose glucocorticoid therapy may be effective in treating some cases of lymphocytic hypophysitis when neurologic sequelae are present.
Diabetes Insipidus
Arginine vasopressin-antidiuretic hormone (AVP-ADH) is a cyclic nonapeptide secreted by the axonal terminals of the neurohypophysis emanating from neurosecretory neurons located in the supraoptic and paraventricular nuclei of the hypothalamus. Blood osmolality is carefully monitored by sensitive osmoreceptors in the anterior hypothalamus. AVP-ADH is released in response to increasing osmotic pressures or decreasing hydrostatic pressures, and acts on the kidney to increase water retention. This system is designed to adjust blood osmolality over a relatively narrow range (±1.8%), with a mean of 285 mOsm/kg in nonpregnant women. Pregnancy is associated with a decrease in plasma osmolality of approximately 9 to 10 mOsm/kg, which is evident early in the first trimester and persists throughout gestation and appears to mirror changes in maternal hCG levels. However, circulating AVP-ADH levels do not change in pregnancy. These data suggest that pregnancy is associated with a modest resetting of the osmostat, leading to a 9 to 10 mOsm/kg decrease in the osmotic threshold for AVP-ADH release.
DI involves the inappropriate loss of water resulting from failure of adequate tubular reabsorption by the kidney. The condition is characterized by polyuria (defined as more than 3 L of urine in 24 hours), polydipsia, and plasma hyperosmolarity. The causes of DI can be divided into two groups: central and peripheral.
Central (hypothalamic) DI refers to lesions of the hypothalamus or posterior pituitary that lead to inadequate production of AVP-ADH. The differential diagnosis of central DI includes pituitary surgery, trauma, infection, and infiltration of the neurohypophysis by tumors or inflammatory cells. Central DI is typically characterized by the acute onset of massive polyuria of 4 to 15 L per day. Peripheral (nephrogenic) DI refers to peripheral resistance to AVP-ADH action. Measurement of plasma AVP-ADH levels may be able to distinguish these two groups (levels are low in central DI and elevated in nephrogenic DI).
Transient nephrogenic DI can occur in pregnancy, usually in association with preeclampsia, HELLP (hemolysis, elevated liver enzymes, low platelet count) syndrome, or acute fatty liver of pregnancy. High levels of placental vasopressinase may contribute to pregnancy-associated DI by degrading endogenous AVP-ADH. This increase in vasopressinase activity may also cause women with partial hypothalamic AVP-ADH deficiency to develop overt DI in pregnancy. D-arginine vasopressin (DDAVP) is resistant to degradation by placental vasopressinase. As such, DDAVP may be more effective than native AVP-ADH in the treatment of women with DI. In most cases, DI improves after delivery.
DI is a rare disease in pregnancy. If suspected, the diagnosis of DI should be confirmed by performing a water-deprivation test. After an overnight fast, the patient is denied water until 3% of body weight is lost or urine osmolarity shows no increment in three successive hourly specimens. In women with DI, urine osmolarity remains low while plasma osmolarity increases significantly. This test is best performed by an endocrinologist because of the risks associated with dehydration and hypernatremia. To help identify the cause, 10 µg of DDAVP can be administered immediately after the completion of the water-deprivation test. In women with central DI, there will be a decrease in urine output and an increase in urine osmolarity. In women with nephrogenic DI, on the other hand, there will be only a minimal change in urine output and osmolarity.
Disorders of Thyroid Function
- ◆
Pregnancy-related changes in thyroid function include (1) relative maternal iodide deficiency and resultant increase in volume of the thyroid gland by 10% to 20%; (2) increase in maternal serum concentration of TBG; (3) increased circulating total T 3 and T 4 . Data are conflicting regarding changes in free T 4 throughout pregnancy, and some free T 4 immunoassays may be unreliable in pregnancy, due to an increase in TBG concentration and lower albumin concentrations; (4) decreased serum TSH concentration in the first trimester.
- ◆
The fetal thyroid gland and fetal pituitary-thyroid axis becomes functional late in the first trimester. (Fetal thyroid begins to concentrate iodine at 9 to 10 weeks’ gestation; TBG and T 4 are first detected in fetal serum at 10 to 12 weeks’ gestation.) The majority of fetal thyroid hormone synthesis occurs after the 18th to 20th week of gestation, and fetal thyroid secretion increases gradually thereafter, reaching a plateau at 35 to 37 weeks.
- ◆
Maternal hyperthyroidism in pregnancy is associated with adverse maternal and fetal outcomes, and the mainstay of therapy remains thionamide treatment (with PTU or methimazole). Graves disease is the most common cause of maternal hyperthyroidism in pregnancy, accounting for 95% of all cases. Because IgG antibodies in Graves disease cross the placenta, the fetus is at risk of immune-mediated thyroid dysfunction. This fetal risk remains in women with Graves disease who have undergone thyroid ablation prior to pregnancy.
- ◆
Maternal hypothyroidism in pregnancy is also associated with adverse maternal and fetal outcomes, and early identification and treatment with levothyroxine or other thyroid hormone can ameliorate maternal and fetal risks. In developed countries, chronic autoimmune thyroiditis (Hashimoto disease) is the most common cause of maternal hypothyroidism in pregnancy, while worldwide the most common cause is iodine deficiency.
- ◆
Treatment of subclinical hypothyroidism (SCH; elevated TSH with normal free T 4 levels) and isolated maternal hypothyroxinemia has not been demonstrated to improve cognitive outcomes in offspring. The impact of treatment on adverse obstetric outcomes remains unclear, and data are conflicting in this regard.
For reasons that are not fully understood, thyroid disease is 5 to 10 times more common in females than in males at all ages. Moreover, many of these conditions are autoimmune in nature, with a peak incidence during the childbearing years. For these reasons, thyroid disease is one of the most common endocrine diseases affecting women of reproductive age, and despite the adverse effect of thyroid disease on fertility, such disorders are commonly encountered in pregnancy. Thyroid disease may also present for the first time during pregnancy. Pregnancy may complicate the management of functional thyroid disorders by affecting their clinical manifestations and limiting the approaches commonly used for diagnosis and treatment. The approach to other thyroid conditions like nodular disease must also be modified during pregnancy because of concern for the safety of the fetus.
Thyroid Physiology
Normal Thyroid Function
Thyroid hormone production is dependent in large part on the supply of iodine, which is derived solely from dietary sources and actively transported into the thyroid gland. The functional unit of the thyroid gland is the thyroid follicle, which is composed of a spherical alignment of cuboid epithelial cells surrounding a core of colloid. Colloid consists primarily of thyroglobulin, which provides tyrosine residues for serial iodination that results, through a complex series of biochemical and biophysical alterations, in the production of the thyroid hormones T 4 and T 3 . The thyroid gland is responsible for the production of all circulating T 4 and approximately 20 % of T 3 . Most of the body’s supply of T 3 results from peripheral conversion of T 4 through a variety of tissue-specific deiodinase enzymes.
The thyroid hormones, T 4 and T 3 —whose total serum concentrations in nonpregnant women are approximately 4 to 12 µg/dL and 90 to 200 ng/mL, respectively ( Table 27.6 )—circulate in a mostly bound form, such that less than 1% circulates as free hormone. Thyroid hormone is bound primarily to a specific serum-binding protein known as TBG, with lesser amounts bound to albumin and prealbumin. As in most endocrine systems, it is the free fraction of these hormones, not the total concentration, that are physiologically important.
| Thyroid Function Test | Units | Normal | Comparison to Nonpregnant Values | Hyperthyroidism | Hypothyroidism | |
|---|---|---|---|---|---|---|
| Nonpregnant Values | Pregnant Values | |||||
| Thyroid-stimulating hormone (TSH) | mIU/L | 0.4–4.0 | First trimester: 0.1–2.5 * Second trimester: 0.2–3.0 * Third trimester: 0.3–3.0 * | Decreased (largest decrease in first trimester) | Markedly decreased | Markedly increased |
| Thyroid-binding globulin (TBG) | mg/L | 11–21 | 23–25 | Increased | No change | No change |
| Total levothyroxine (T 4 ) | µg/dL | 3.9–11.6 | 10.7–11.5 | Increased | Increased | Decreased |
| Free levothyroxine (T 4 ) | ng/dL | 0.8–2.0 | Trimester-specific and method-specific ranges should be used | Decreased or no change | Increased | Decreased |
| Total L-triiodothyronine (T 3 ) | ng/dL | 91–208 | 205–233 | Increased | Normal to increased | Normal to decreased |
| Free L-triiodothyronine (T 3 ) | pg/dL | 190–710 | 250–330 | No change | Increased | Decreased |
* For use if trimester-specific ranges for thyroid-stimulating hormone (TSH) are not available from the laboratory.
Regulation of Thyroid Hormone Secretion
Under normal circumstances, the circulating concentration of T 4 , the most abundant and commonly measured thyroid hormone, is maintained within a narrow range that varies little from day to day. The follicular cell within the thyroid gland is responsible for the uptake of inorganic iodide from the circulation, its organification into iodinated thyronine compounds, storage of thyroid prohormone in the form of thyroglobulin, and reuptake of formed thyroid hormone and its ultimate release into the systemic circulation.
Follicular cell activity is under the direct control of the hypothalamic-pituitary-thyroid axis ( Fig. 27.13 ). The hypothalamus produces the tripeptide TRH, which enters the portal circulation of the infundibular stalk and travels to the anterior lobe of the pituitary, where it stimulates specific cells (thyrotropes) to produce TSH. TSH secretion varies diurnally with a peak secretion occurring between 11 p.m. and 4 a.m. TSH enters the systemic circulation and interacts with specific heptahelical, G-protein-coupled receptors on the surface of thyroid follicular cells, which triggers a series of signal transduction cascades culminating in the synthesis and release of thyroid hormones. Through a classical endocrine negative feedback loop, decreased circulating levels of thyroid hormones lead to an increase in TRH and TSH secretion (see Fig. 27.13 ), which in turn leads to increased thyroid growth and activity.
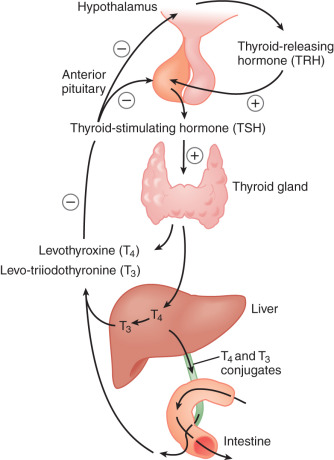
Physiological Role of Thyroid Hormone
The precise role of thyroid hormone remains incompletely understood, although it clearly interacts with numerous biological systems. This fact is underscored by the complex series of symptoms and signs that are evident in patients with thyroid dysfunction ( Fig. 27.14 ). At a cellular level, the active hormone (T 3 ) is transported into cells where it interacts with specific nuclear receptors. The T 3 -receptor complex binds to specific thyroid hormone response elements within the promoter sequences of target genes and functions as a transcription factor, working along with other nuclear proteins to regulate gene expression. In addition to these genomic effects, thyroid hormone also appears to have important extranuclear actions. These actions include regulation of deiodinase activity and, possibly, mitochondrial function.
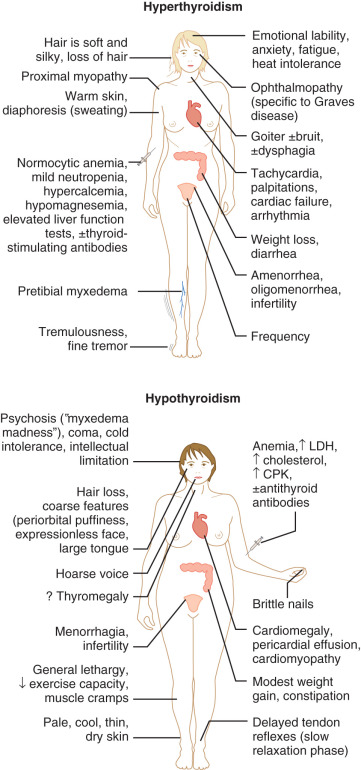
Effect of Pregnancy on Thyroid Function
Maternal Thyroid Function in Pregnancy
In pregnancy, renal clearance of iodide increases (because of an increase in the glomerular filtration rate) and substantial amounts of iodide and iodothyronines are transferred to the fetus. As pregnancy progresses and fetal thyroid hormone production increases, the fetus needs increasing amounts of iodide. To meet this demand, the placenta is able to rapidly and efficiently transport available iodide from the maternal to the fetal circulation. The placenta is also capable of mono-deiodination of iodothyronines, thereby making more iodide available for transport. The net result of these pregnancy-related physiological alterations is a decrease in the circulating concentration of inorganic iodide during pregnancy and a resultant increase in volume of the thyroid gland by 10% to 20% during pregnancy. In light of this relative iodide deficiency during pregnancy, the recommended daily intake of iodine is increased from a baseline of 100 to 150 µg/day to approximately 250 µg/day. To achieve this daily dose, the American Thyroid Association (ATA) recommends that pregnant and lactating women supplement dietary iodine intake with a prenatal vitamin including 150 µg/day of iodine; this is the dose included in the majority of prenatal vitamins in the United States.
Serum concentrations of TBG increase in pregnant women by 75% to 100% (the T 4 resin uptake decreases proportionally). Moreover, much of the increase in circulating TBG levels occurs during the first trimester and results from the effects of the hyperestrogenemic state on hepatocytes, with stimulation of TBG synthesis and reduced hepatic clearance due to estrogen-induced TBG sialylation. The concentration of TBG plateaus at around 12 to 14 weeks’ gestation and is associated with a concomitant increase in circulating total thyroid hormone concentrations ( Fig. 27.15 ).
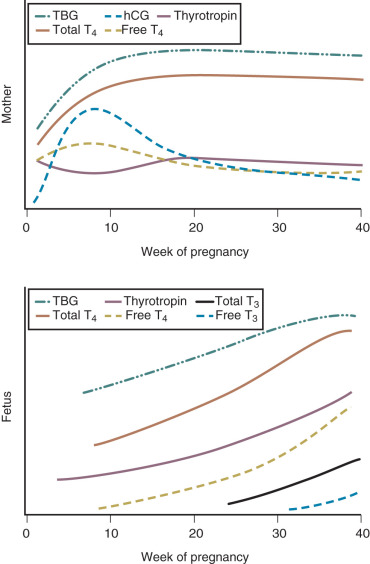
Indeed, mean concentrations of both total T 4 and T 3 in the maternal circulation increase by 10% to 30% in most longitudinal studies, usually into a range that is considered elevated in the general population. In the first trimester, the increase in total T 4 exceeds the rise in TBG, resulting in a slight increase in free T 4 , although free hormone levels typically return to normal by the early second trimester (see Fig. 27.15 ). However, these changes are so subtle that serum-free T 4 concentrations in most pregnant women remain within the normal range for nonpregnant women. While some studies report a substantial decrease in serum free T 4 with progression of gestation, some report no change in T 4 concentrations as gestation progresses. The negative-feedback control system of the hypothalamic-pituitary-thyroid axis functions normally in pregnant women. Some free T 4 (and possibly T 3 ) immunoassays may be unreliable in pregnancy , due to an increase in TBG concentration and lower albumin concentrations.
While the International Federation of Clinical Chemistry and Laboratory Medicine recommends using an isotope dilution-liquid chromatography/tandem mass spectrometry (LC/MS/MS) method for measuring T 4 in the dialysate from equilibrium dialysis of serum to obtain a reference measurement for serum free T 4 , this assay technology is not currently widely available due to high cost.
The ATA thus recommends the use of online extraction/LC/MS/MS to measure FT 4 in the dialysate or ultrafiltrate of serum samples. If this method is unavailable, they recommend method-specific and trimester-specific reference ranges for serum free T 4 . If these adjusted reference ranges are unavailable, or if free T 4 measurements are discordant with TSH measurements, consideration can be given to using adjusted serum total T 4 measurements to assess thyroid function. Total T 4 and T 3 levels in pregnancy are typically 1.5-fold higher than in nonpregnant women, so an adjusted reference range should be used.
The pregnancy-specific glycoprotein hormone hCG is structurally similar to TSH and has some weak thyrotropic activity, which is estimated at approximately 0.025% that of TSH. The production of hCG begins during the first week after fertilization and is highest near the end of the first trimester, after which it declines. This increase causes a transient increase in serum free T 4 concentrations, which in turn decreases serum TSH concentrations during the first trimester (see Fig. 27.15 ). Because hCG concentrations are higher in multiple gestations compared with singleton pregnancies, the suppression of serum TSH concentrations is more marked in multiple gestations. This cross-reactivity only becomes clinically significant if circulating levels of hCG are markedly elevated, such as those seen in complete molar pregnancies.
Fetal Thyroid Function in Pregnancy
The fetal thyroid gland and pituitary-thyroid axis becomes functional late in the first trimester. Before that time, any thyroid hormone in the fetus must come from the maternal circulation. By 9 to 10 weeks of gestation, the fetal thyroid begins to concentrate iodine, thyroid follicles become visible, and T 4 synthesis can be demonstrated. TBG and T 4 are first detected in fetal serum at approximately 10 to 12 weeks of gestation, although the majority of fetal thyroid hormone synthesis occurs after the 18th to 20th week of gestation. From this point forward, fetal thyroid secretion increases gradually, reaching a plateau at 35 to 37 weeks. At term, fetal serum TSH concentrations are higher, free T 4 concentrations are slightly lower, and T 3 concentrations are one-half those in the maternal circulation. The progressive increase in serum TBG concentrations with increasing gestational age presumably reflects maturation of the fetal liver and its responsiveness to estrogen stimulation. Increases in pituitary and serum concentrations of TSH during the second trimester coincide with the development of the hypothalamic-pituitary portal circulation, which facilitates the regulation of pituitary TSH secretion by hypothalamic TRH. The increased secretion of TRH despite higher serum-free T 4 concentrations implies immaturity of the negative-feedback system that regulates the secretion of TSH and TRH in utero.
Transplacental passage of thyroid hormones (T 4 and T 3 ) from the mother to the fetus does occur but is minimal, estimated at less than 0.1%. This is likely due to the large amount of type III deiodinase enzyme in the placenta, which serves to maintain low serum T 3 concentrations in the fetus while protecting decidual cells from hypothyroidism. As such, tests of fetal thyroid function—although rarely, if ever, indicated in clinical practice—accurately reflect functioning of the fetal thyroid and are largely unrelated to maternal thyroid status. That said, however, in neonates with congenital hypothyroidism, enough maternal thyroid hormone is able to cross the placenta to prevent the overt stigmata of hypothyroidism at birth and maintain cord blood thyroid hormone levels at approximately 25% to 50% of normal. Iodine, TRH, and TSH receptor immunoglobulins do cross the placenta, as does TSH, but to a far lesser extent. Fetal thyroid function is completely dependent on the supply of iodine from the mother. Normal levels of thyroid hormone are critical for neuronal migration and myelination of the fetal brain. Thus maternal and fetal iodine deficiency in pregnancy has adverse effects on the neurocognitive function of offspring. Children born to severely iodine-deficient mothers may exhibit profound mental retardation, deaf-mutism, and motor rigidity, a constellation of symptoms known as cretinism.
Thyroid hormone is also present in measurable quantities in amniotic fluid. At term, total T 4 concentrations in the amniotic fluid are about 0.6 µg/dL, much lower than in maternal or fetal serum. Because protein and TBG concentrations are low in amniotic fluid, however, the free T 4 and T 3 concentrations are slightly higher than in maternal or fetal serum. The source of this thyroid hormone is not known, but studies in fetuses with congenital hypothyroidism suggest that it may come from the maternal circulation. Late in gestation, fetal swallowing appears to allow for the transfer of thyroid hormones from amniotic fluid to the fetal circulation. The physiological role of thyroid hormone in the amniotic fluid is not known.
Functional Thyroid Disorders in Pregnancy
Screening for Thyroid Disorders in Pregnancy
Data from the US population suggest that 2% to 3% of pregnant women will have an elevated TSH level at the time of routine screening. Of screened women, 0.3% to 0.5% will have overt hypothyroidism (elevated TSH and low free T 4 , or a TSH level of 10 mIU/L or greater, regardless of free T 4 levels), and 2% to 2.5% will have SCH (TSH levels above the 97.5th percentile for gestational age with normal free T 4 concentration). Hyperthyroidism is less common, and occurs in only 0.1% to 0.4% of pregnant women. Overt thyroid dysfunction in pregnancy has consistently been associated with increased risk of adverse pregnancy outcomes and detrimental effects on fetal neurocognitive development (see the sections on “ Maternal Thyrotoxicosis ” and “ Maternal Hypothyroidism ”). To date, there is no consistent evidence that screening and treatment for asymptomatic hypothyroidism will abrogate the posited association between SCH and neurocognitive impairment in offspring. Subclinical hyperthyroidism has not been associated with adverse maternal or fetal outcomes.
While two decision analyses found that universal screening for thyroid dysfunction in pregnancy is cost-effective, both models assumed that treatment of SCH in pregnancy would increase offspring IQ, which has not been demonstrated in large randomized clinical trials. Although large studies of a targeted case-finding strategy versus universal screening to identify women with hypothyroidism in pregnancy have demonstrated that a targeted case-finding approach may miss as many as 30% to 55% of women with thyroid abnormalities, universal screening has not been shown to result in improved population outcomes, and treatment of pregnant women with SCH has not been shown to result in improved childhood cognitive function. Given these data, ACOG, the ATA, and the Endocrine Society recommend a targeted case-finding approach, rather than universal first-trimester screening of all pregnant women for SCH. The ATA’s suggested criteria for whom to screen for thyroid dysfunction in pregnancy are summarized in Box 27.4 . TSH measurement in the first trimester should be performed in these women, with free T 4 measurement if TSH is greater than 2.5 mIU/L. Screening is not indicated in asymptomatic pregnant women who have a mildly enlarged thyroid, in the absence of goiter.
Family or personal history of thyroid disease
Symptoms suggestive of thyroid dysfunction (see Fig. 27.14 )
From an area with known moderate to severe iodine insufficiency
Type 1 diabetes or other autoimmune disorders frequently associated with autoimmune thyroid dysfunction (vitiligo, adrenal insufficiency, hypoparathyroidism, atrophic gastritis, pernicious anemia, systemic sclerosis, systemic lupus erythematosus, Sjögren syndrome)
Known thyroid peroxidase antibodies
History of head or neck radiation
History of preterm delivery or recurrent miscarriage
Morbid obesity (BMI ≥ 40 kg/m 2 )
Infertility
Age > 30 years
Use of amiodarone or lithium
Recent administration of iodinated radiologic contrast
BMI , Body mass index.
Maternal Thyrotoxicosis
Thyrotoxicosis is the clinical and biochemical state that results from an excess production of and exposure to thyroid hormone from any cause. In contrast, hyperthyroidism refers to thyrotoxicosis caused by hyperfunctioning of the thyroid gland. Hyperthyroidism occurs in 0.05% to 0.2% of pregnancies. Graves disease is the most common cause of maternal hyperthyroidism in pregnancy, accounting for 95% of cases. Other causes of thyrotoxicosis in pregnancy are summarized in Box 27.5 . Of note, gestational hyperthyroidism, defined as “transient hyperthyroidism limited to the first half of pregnancy [and] characterized by elevated free T 4 or adjusted total T 4 and suppressed or undetectable serum TSH, in the absence of serum markers of thyroid autoimmunity,” is a frequent cause of thyrotoxicosis in pregnancy. Gestational hyperthyroidism occurs in one to 3% of pregnancies, and may be associated with hyperemesis gravidarum. Appropriate management includes supportive therapy and management of dehydration, if necessary. Antithyroid medications are not recommended as part of the standard management of gestational hyperthyroidism. While gestational hyperthyroidism occurs in 1% to 3% of pregnancies, hyperemesis gravidarum, a syndrome of nausea and vomiting associated with weight loss of 5% or more in early pregnancy, occurs in 0.5 to 10 per 1000 pregnancies. Hyperemesis gravidarum is characterized by higher serum hCG and estradiol concentrations than in nonaffected pregnant women, and hCG has more thyroid-stimulating activity in women with hyperemesis gravidarum.
- •
Graves disease: The most common cause of maternal thyrotoxicosis in pregnancy (95%). Results from the circulating thyroid-stimulating immunoglobulin G autoantibodies, which can cross the placenta leading to fetal thyroid dysfunction
- •
Thyroiditis (silent/postpartum [lymphocytic], subacute [granulomatous]; suppurative [bacterial]): Characterized by hyperthyroidism and the presence of a large, palpable thyroid gland. An acute enlarged, painful, and tender thyroid is suggestive of subacute (De Quervain’s) thyroiditis.
- •
Toxic multinodular goiter
- •
Solitary toxic nodule (also referred to as a hyperfunctioning thyroid adenoma)
- •
Gestational trophoblastic neoplasia: Including hydatidiform mole and choriocarcinoma. Probably secondary to elevated levels of hCG
- •
Struma ovarii: Refers to thyroid tissue in a mature ovarian teratoma
- •
Exogenous thyroid hormone: Most commonly due to inadvertent ingestion of thyroid hormone
- •
Iodine-induced thyrotoxicosis
- •
TSH-secreting pituitary adenoma
- •
Gestational hyperthyroidism: Transient hyperthyroidism limited to the first half of pregnancy, frequently associated with elevated levels of hCG. Hyperemesis gravidarum is often coexistent. Typically self-limited, supportive care is indicated
- •
Hyperemesis gravidarum: Characteristic symptoms and signs of hyperthyroidism are often absent
- •
Familial gestational hyperthyroidism: TSH receptor mutation leading to functional hypersensitivity to hCG
hCG , Human chorionic gonadotropin; TSH , thyroid-stimulating hormone.
Numerous drugs are known to interfere with thyroid hormone synthesis and metabolism ( Box 27.6 ). Symptoms and signs may suggest the etiology of thyrotoxicosis in pregnancy (see Fig. 27.14 ). For example, endocrine ophthalmopathy (lid lag, lid retraction) and dermopathy (localized or pretibial edema) are clinical signs that are specific to Graves disease. However, as in nonpregnant patients, the confirmation of maternal hyperthyroidism in pregnancy requires thyroid function testing (see Table 27.6 ). Measurement of TSH receptor antibodies and total T 3 may be helpful in trying to distinguish between Graves disease and gestational hyperthyroidism. Radioactive iodine scanning or radioiodine uptake determination should not be performed in pregnancy. There is not sufficient evidence to recommend for or against the use of thyroid ultrasound in distinguishing between possible etiologies of hyperthyroidism in pregnancy.
Inhibition of Thyroid Hormone Synthesis by Thyroid Gland
Iodine, sulfonylureas, lithium
Increase in TSH
Iodine, cimetidine, dopamine agonists, lithium
Decrease in TSH
Glucocorticoids, dopamine agonists, somatostatin
Inhibition of Thyroid Hormone Binding to TBG
Phenytoin, diazepam, sulfonylureas, furosemide, salicylates
Inhibition of Conversion of T 4 to T 3 in Peripheral Tissues (Liver)
Glucocorticoids, PTU, ipodate, propranolol, amiodarone
Inhibition of Gastrointestinal Resorption of Thyroid Hormones
Cholestyramine, cholestipol, ferrous sulfate
PTU , Propylthiouracil; TBG , thyroid-binding globulin; TSH , thyroid-stimulating hormone.
As compared with well-controlled disease, inadequately treated maternal hyperthyroidism is associated with infertility and adverse perinatal outcome. Maternal complications in pregnancy include an increased risk of preeclampsia, cardiac failure, thyroid storm, and possibly spontaneous pregnancy loss. Fetal and neonatal risks that are increased in the setting of poorly controlled hyperthyroidism include preterm delivery, low birth weight, IUGR, stillbirth, central hypothyroidism, and increased perinatal mortality. Because a large proportion of hyperthyroidism in pregnancy is mediated by IgG antibodies that cross the placenta (Graves disease and chronic autoimmune thyroiditis), the fetus is at risk of immune-mediated thyroid dysfunction. It is important to remember this is also true in women with a history of Graves disease treated with thyroidectomy or radioactive iodine, and in fact these women’s fetuses may be at greater risk of fetal hyperthyroidism because the mothers are not on thionamide treatment. Fetal sinus tachycardia (>160 beats per minute persistent for over 10 minutes) is a sensitive index of fetal hyperthyroidism. Only 1% to 5% of neonates born to women with poorly controlled thyrotoxicosis will develop transient hyperthyroidism or neonatal Graves disease caused by the transplacental passage of maternal antithyroid antibodies. In addition to fetal tachycardia, other signs of fetal hyperthyroidism include IUGR, presence of fetal goiter, and craniosynostosis or accelerated bone maturation. Congestive heart failure and fetal hydrops may occur in severe cases. Fetuses of women with Graves disease are at risk not only for fetal and neonatal hyperthyroidism, but also for fetal and neonatal hypothyroidism due to overtreatment with antithyroid drugs, and central hypothyroidism. High titers of serum thyroid receptor antibodies in the late second and early third trimester are a risk factor for fetal or neonatal hyperthyroidism, and thus determination of maternal serum thyroid receptor antibodies is recommended at 20 to 24 weeks’ gestation in women with a history of Graves disease. Fetal surveillance with serial ultrasounds is recommended in pregnant women with Graves disease, uncontrolled hyperthyroidism, or those with thyroid receptor antibody levels greater than two to three times the upper limit of normal.
To minimize complications, hyperthyroidism is best diagnosed and treated prior to conception, and the use of contraception is advisable until a euthyroid state is achieved. If a patient has high TSH receptor antibody titers and is planning pregnancy in the next 2 years, surgical therapy may be considered. TSH receptor antibody titers tend to increase and remain elevated for many months after 131 I therapy. If 131 I ablative therapy is performed, pregnancy should be delayed for 6 months after thyroid ablation. The goal of therapy during pregnancy is to control thyrotoxicosis while avoiding fetal and transient neonatal hypothyroidism. The mainstay of treatment for hyperthyroidism in pregnancy is thionamide drugs, specifically propylthiouracil (PTU) and methimazole (the active metabolite of carbimazole), which decrease thyroid hormone synthesis by blocking the organification of iodide. PTU also reduces the peripheral conversion of T 4 to T 3 , and may therefore have a quicker suppressive effect than methimazole.
Traditionally, PTU has been preferred in pregnant patients because methimazole was believed to pass the placenta more easily and was associated with methimazole embryopathy (including choanal or esophageal atresia, tracheoesophageal fistula, patent vitellointestinal duct omphalocele, omphalomesenteric duct anomaly, and dysmorphic facies) and fetal aplasia cutis congenita, a rare congenital skin defect of the scalp. Congenital malformations have also been reported in conjunction with PTU use, however, including urinary tract malformations, limb/hand/foot malformations, omphalocele/umbilical cord abnormalities, and malformations in the face and neck region. The preponderance of available evidence suggests that carbimazole/methimazole and PTU are both associated with an increased risk for birth defects, with most studies suggesting an increased risk of congenital anomalies with carbimazole/methimazole compared with PTU. Some of these data should be interpreted with caution, however, given that the registry-based cohort data may not take into account historical prescribing practices (e.g., carbimazole was historically prescribed far more frequently in the UK than PTU).
Despite concern about theoretical risk of methimazole embryopathy and fetal aplasia cutis, PTU treatment is clearly associated with an increased risk for fulminant hepatotoxicity and agranulocytosis. Hepatotoxicity may occur at any time during PTU treatment, and there are no data indicating that monitoring of liver function tests is effective in preventing fulminant hepatotoxicity. Thus an advisory committee to the US Food and Drug Administration recommended limiting the use of PTU to the first trimester of pregnancy. PTU treatment is usually initiated at 50 to 300 mg daily in divided doses (the lowest dose is preferable to minimize the risk of fetal hypothyroidism). Following the first trimester, consideration should be given to transitioning to methimazole, usually initiated at 5 to 15 mg daily. Methimazole is approximately 20 to 30 times as potent as PTU; thus 300 mg of PTU is approximately equivalent to 10 or 15 mg of methimazole. The goal is to use the least amount of drug to maintain maternal free T 4 at the upper limits of normal. TSH and T 4 levels should be checked monthly and treatment adjusted accordingly, as maternal overtreatment and fetal hypothyroidism are possible. Clinicians should remember that stored hormone may not be depleted for 3 to 4 weeks, therefore delaying a clinical response. Complete blood counts should be monitored monthly because of the risk of drug-induced agranulocytosis.
Radioactive iodine ( 131 I) administration to ablate the thyroid gland is absolutely contraindicated in pregnancy. Moreover, breastfeeding should be avoided for at least 120 days after 131 I treatment. Surgery is best avoided, but may be performed in the second trimester if indicated for failed medical therapy.
Subclinical hyperthyroidism, defined as low TSH with normal free T 4 and T 3 in an asymptomatic patient, is not associated with adverse perinatal outcome. Long-term subclinical hyperthyroidism is associated with osteoporosis, atrial fibrillation, and increased mortality, and as such should be treated. However, there are few data on the appropriate course of action during pregnancy. Many cases resolve spontaneously. The presence of suppressed serum TSH in the first trimester (TSH < 0.1 mIU/L) should be investigated with a history and physical examination, as well as free T 4 measurements in all patients.
Thyroid Storm
Thyroid storm (thyrotoxic crisis) is a medical emergency characterized by a severe acute exacerbation of the signs and symptoms of hyperthyroidism. It is a rare complication, occurring in approximately 1% of pregnant patients with hyperthyroidism, but is associated with a high rate of maternal mortality and morbidity. Thyroid storm is diagnosed by a combination of the following symptoms and signs in patients with thyrotoxicosis: fever, tachycardia out of proportion to the fever, altered mental status (restlessness, nervousness, confusion, seizures), diarrhea, vomiting, and cardiac arrhythmia. An inciting event (infection, surgery, labor, and delivery) can be identified in many instances. The diagnosis can be difficult to make, however, and requires expeditious treatment to avoid severe consequences like shock, stupor, coma, and death.
If thyroid storm is suspected, serum TSH and free T 4 and T 3 levels should be evaluated to help confirm the diagnosis. Patients with thyroid storm will have suppressed TSH and high free T 4 and/or T 3 concentrations. Laboratory derangements may be similar in magnitude to those of patients with uncomplicated overt hyperthyroidism, so the degree of hyperthyroidism does not correspond to the likelihood of thyroid storm, if clinical findings are suggestive. If the clinical index of suspicion is high, treatment should not be withheld or delayed pending the results of the biochemical tests. The treatment of thyroid storm is summarized in Box 27.7 . The goals of treatment are to
- •
Reduce synthesis and release of hormone from the thyroid gland (using thionamide like PTU or methimazole, supplemental iodide, and glucocorticoids).
- •
Block the peripheral actions of thyroid hormones (using glucocorticoids, PTU, and high-dose beta blockers).
- •
Reduce enterohepatic recycling of thyroid hormone using bile acid sequestrants.
- •
Treat complications and support physiologic functions (supplemental oxygen, fluid, and caloric replacement).
- •
Identify and treat precipitating events (such as hypoglycemia, thromboembolic events, and DKA).
- •
Propylthiouracil (PTU) 600–800 mg orally stat, then 150–200 mg orally every 4–6 h. If oral administration is not possible, consider nasogastric tube administration. Methimazole rectal suppositories or PTU enema/suppositories can also be prepared by most hospital pharmacies if ordered in advance.
- •
Starting 1–2 h after PTU, administer saturated solution of potassium iodide (SSKI) , 2–5 drops orally every 6 h, or sodium iodide , 0.5–1.0 g intravenously every 8 h, or Lugol solution , 8–10 drops every 8 h, or lithium carbonate , 300 mg orally every 6 h.
- •
Dexamethazone , 2 mg intravenously or intramuscularly every 6 h for four doses. (Alternative: hydrocortisone 100 mg IV q 8 h.)
- •
Propranolol , 60–80 mg orally every 4–6 h or 1–2 mg intravenously every 5 min for a total of 6 mg, then 1–10 mg intravenously every 4 h. If the patient has a history of severe bronchospasm, reserpine (1–5 mg intramuscularly every 4–6 h), guanethidine (1 mg/kg orally every 12 h), or diltiazem (60 mg orally every 6–8 h) should be given.
- •
Phenobarbital , 30–60 mg orally every 6–8 h as needed for extreme restlessness.
- •
Cholestyramine , 4 g orally four times daily to decrease enterohepatic recycling of thyroid hormone.
- •
Aspirin should not be used to treat fever, as it can increased serum free T 4 /T 3 concentrations by interfering with protein binding.
As with other acute maternal illnesses, fetal well-being should be evaluated and consideration given to delivery, if appropriate.
Maternal Hypothyroidism
Hypothyroidism is caused by inadequate thyroid hormone production. It complicates 0.3% to 0.5% of all pregnancies but is more common in women with other autoimmune diseases, such as type 1 diabetes. The classic signs and symptoms of hypothyroidism (summarized in Fig. 27.14 ) may suggest the diagnosis. Again, however, thyroid function testing is required for a definitive diagnosis (see Table 27.6 ). The causes of hypothyroidism in pregnancy are summarized in Box 27.8 . In developed countries, chronic autoimmune thyroiditis (Hashimoto disease) is the most common cause. Worldwide, however, the most common cause of hypothyroidism is iodine deficiency. Women previously treated for Graves disease (by radioactive iodine or surgery) may manifest with posttherapy hypothyroidism. Although such women may themselves be asymptomatic, their fetuses remain at risk for thyroid dysfunction, as circulating antithyroid antibodies are still present. In women with preexisting Hashimoto disease, pregnancy may actually result in a transient improvement of symptoms.
Primary Hypothyroidism
- •
Iodine deficiency (the most common cause of hypothyroidism worldwide)
- •
Chronic autoimmune thyroiditis (Hashimoto): Characterized by hypothyroidism, a firm goiter, and the presence of circulating antithyroglobulin and/or antimicrosomal autoantibodies
- •
Silent/postpartum thyroiditis (hypothyroid phase)
- •
Prior treatment for hyperthyroidism (includes women previously treated with radioactive iodine or surgery [thyroidectomy] leading to posttherapy hypothyroidism)
- •
Prior high-dose external beam neck irradiation
- •
Infectious (suppurative) thyroiditis: Characterized by fever and a painful, swollen thyroid gland. Common infections include Staphylococcus aureus, S. haemolyticus , and fungi.
- •
Subacute thyroiditis (hypothyroid phase): Similar to suppurative thyroiditis, but it is usually the result of a viral infection and is self-limiting
- •
Thyroid agenesis/dysgenesis
- •
Drug-induced hypothyroidism
- •
Dietary goitrogens (includes such drugs as thionamides and lithium)
- •
Organification enzyme defects
Secondary Hypothyroidism
- •
Pituitary adenoma
- •
Pituitary necrosis/hemorrhage
- •
Lymphocytic hypophysitis
- •
Central nervous system sarcoidosis
- •
Prior hypophysectomy
- •
Cranial irradiation
- •
Suprasellar/parasellar
- •
Traumatic injury to pituitary/hypothalamus
Untreated or inadequately treated maternal hypothyroidism in pregnancy is associated with an increased risk of adverse pregnancy outcome, including preeclampsia, low birth weight, placental abruption, preterm birth, and stillbirth. It is not clear whether untreated hypothyroidism is a risk factor for IUGR, independent of other complications. Thyroid hormones also have important roles in embryogenesis and fetal maturation. Maternal hypothyroxinemia is associated with neonatal hypothyroidism and with defects in IQ and long-term neurologic function in the offspring. Women with iodine-deficient hypothyroidism are at particularly high risk of having a child with congenital cretinism (growth failure, mental retardation, and other neuropsychological deficits).
Early diagnosis and treatment of maternal hypothyroidism is essential to avoid antepartum pregnancy complications and impaired neonatal and childhood development. Indeed, in an iodine-deficient population, treatment with iodine in the first and second trimesters of pregnancy has been shown to significantly reduce the incidence of congenital cretinism. With the advent of routine newborn screening for congenital hypothyroidism, it has become clear that size, weight, appearance, behavior, extrauterine adaptation, and immediate postnatal development are usually normal in infants with hypothyroidism, even those with thyroid agenesis.
Although untreated hypothyroidism is associated with adverse perinatal outcome, the maternal and fetal consequences of isolated maternal hypothyroxinemia (normal TSH in conjunction with free T 4 concentration in the lower 5th or 10th percentile of the reference range) and SCH during pregnancy are not as clear. One study using stored serum samples of nearly 17,300 pregnant women reported that isolated maternal hypothyroxinemia was not associated with adverse pregnancy outcomes. However, another study using nearly 11,000 stored serum samples from the multicenter, prospective FASTER trial found that women with hypothyroxinemia and normal TSH had 1.6 times the odds of preterm labor, nearly twice the odds of macrosomia, and 1.7 times the odds of gestational diabetes.
The majority of studies suggest that SCH is associated with increased perinatal risk. A retrospective cohort study of more than 17,000 women demonstrated that untreated SCH was associated with a threefold risk of placental abruption and a 1.8-fold risk of preterm birth before 34 weeks, compared with euthyroid controls. SCH in pregnancy has also been associated with a fourfold risk of fetal death, an increased risk for severe preeclampsia, and an increased miscarriage rate, even in thyroid peroxidase antibody (TPOAb)–negative women. A recent systematic review and meta-analysis found that SCH was significantly associated with a higher risk for pregnancy loss (RR 2.01 [1.66 to 2.44]), placental abruption (RR 2.14 [1.23 to 3.70]), premature rupture of membranes (RR1.43 [1.04 to 1.95]), and neonatal death (RR 2.58 [1.41 to 4.73]). However, other large studies have failed to demonstrate an association between SCH and adverse obstetric outcomes, and the value of levothyroxine therapy in preventing adverse outcomes in pregnancy complicated by SCH remains unclear. The only trial to directly address the impact of treatment for SCH randomized more than 4500 pregnant women to either a case-finding group or universal thyroid screening strategy. Negro and colleagues found that the universal screening approach did not result in an overall decrease in adverse perinatal outcomes, but TPOAb-positive women with SCH (TSH > 2.5 mIU/L) who were treated with levothyroxine had a lower risk of adverse obstetric outcomes, including miscarriage, hypertension, preeclampsia, gestational diabetes, placental abruption, cesarean delivery, congestive heart failure, preterm labor, respiratory distress, neonatal intensive care unit admission, low and high birth weight, preterm or very preterm delivery, low Apgar score, and perinatal death. This finding failed to reach statistical significance, however, due to the broad range of outcomes identified as adverse events and the high proportion of adverse outcomes in euthyroid women.
Most controversial is the association between SCH, isolated maternal hypothyroxinemia, and neurologic impairment in the offspring. A large case-control study reported a seven-point reduction in IQ scores in children born to mothers with untreated TSH elevations in pregnancy, compared with children born to euthyroid mothers. Pop et al. reported impaired psychomotor development in offspring born to women with isolated hypothyroxinemia. Similarly, a retrospective cohort study of Chinese women found that SCH, hypothyroxinemia, and elevated TPOAb titers were associated with lower intelligence scores and impaired motor development of offspring at 25 to 30 months. The Generation R study, a prospective, nonrandomized population-based cohort from the Netherlands, found that while SCH was not significantly associated with cognitive outcomes in offspring, both mild (OR 1.44) and severe (OR 1.8) hypothyroxinemia were associated with expressive language delay, and severe maternal hypothyroxinemia predicted a higher risk of nonverbal cognitive delays (OR 2.03).
Other studies, however, have failed to find an association between maternal SCH and cognitive performance in offspring. To date, only two randomized controlled trials have investigated whether levothyroxine therapy for women with either overt or SCH is associated with an improvement in childhood intellectual development. The Controlled Antenatal Thyroid Screening (CATS) study enrolled nearly 22,000 women with singleton pregnancies, and randomized them before 16 weeks’ gestation to screening and treatment for TSH greater than 97.5th percentile and/or free T 4 less than 2.5th percentile, or storage of serum samples until after completion of pregnancy. The CATS study found no significant difference in mean IQ or in the proportion of children with IQ less than 85 at 3 years of age, in children born to 390 treated mothers compared with 404 untreated mothers. The Maternal Fetal Medicine Unit of the National Institutes of Health conducted two multicenter randomized controlled trials in parallel to evaluate the effects of levothyroxine therapy for pregnant women with SCH and isolated hypothyroxinemia. A total of 677 women with SCH and 526 women with hypothyroxinemia were randomized to treatment with levothyroxine or placebo. Maternal treatment with levothyroxine did not result in improved cognitive outcome in offspring (IQ) at 5 years of age. The most recent ATA guidelines recommend levothyroxine therapy for pregnant women with SCH and anti-TPO antibodies, but conclude there is insufficient evidence to recommend for or against treating women with SCH in the absence of TPO antibodies, and do not recommend therapy for isolated hypothyroxinemia in pregnancy.
Levothyroxine is the treatment of choice for pregnant and nonpregnant women with overt hypothyroidism. Treatment should be initiated at an oral dose of 100 to 150 µg daily. TSH levels should be measured serially every 4 weeks, and the dose of levothyroxine adjusted to maintain TSH levels within trimester-specific ranges. As thyroid hormone production is increased during pregnancy, most women will need an increase in their daily dose by approximately 30% to 50% during pregnancy.
Postpartum Thyroiditis
Postpartum thyroiditis is an autoimmune inflammation of the thyroid gland that presents as new-onset, painless hypothyroidism, transient thyrotoxicosis, or thyrotoxicosis followed by hypothyroidism within 1 year postpartum. The condition occurs in approximately 5% (range 4% to 10%) of women without preexisting thyroid disease and may also occur after early pregnancy loss. Thirty-three to 50% of women with antithyroid antibodies in the first trimester will develop postpartum thyroiditis, with higher titers conferring a greater risk. The classic form begins with transient thyrotoxicosis, followed by transient hypothyroidism, with a return to euthyroid state by the end of the first postpartum year. Studies have found that the classic presentation of postpartum thyroiditis is seen in approximately 20% of cases, while the majority of women (44% to 48%) with postpartum thyroiditis have isolated hypothyroidism (with fatigue, weight gain, and depression), and approximately 30% experience isolated thyrotoxicosis (characterized by dizziness, fatigue, weight loss, and palpitations).
The diagnosis of postpartum thyroiditis requires a high clinical index of suspicion. The diagnosis is confirmed by documenting abnormal serum levels of TSH and T 4 in a previously euthyroid patient. Differentiating postpartum thyroiditis from Graves disease can be challenging. Thyroid receptor antibodies are often positive in Graves disease but usually are negative in postpartum thyroiditis, and radioiodine uptake is elevated or normal in Graves disease and low in postpartum thyroiditis. 123 I or technetium scans are preferable to 131 I scans in breastfeeding women, due to the shorter half-life. The need for treatment in women with postpartum thyroiditis is not clear, although it may be warranted to control symptoms. In one prospective study of 605 asymptomatic pregnant and postpartum women, none of the women with thyrotoxicosis and only 40% of women with hypothyroidism required treatment. If treatment is required, it can usually be tapered within 1 year. The majority of studies examining the impact of postpartum thyroiditis on long-term thyroid function have found that between 10% and 20% of women with postpartum thyroiditis will require long-term treatment. A prospective study of 169 women with postpartum thyroiditis, however, reported a much higher rate of 54% of women with persistent hypothyroidism after 1 year. Women with the highest levels of TSH, high TPO antibody titers, multiparous women, older women, and those with a history of miscarriage have the highest risk for developing permanent hypothyroidism.
Both levothyroxine and iodine treatment during pregnancy have been investigated as a potential strategy to prevent postpartum thyroiditis in women with antithyroid antibodies, but neither intervention was effective. A prospective placebo-controlled study evaluating the efficacy of selenium in preventing postpartum thyroiditis in TPOAb-positive women found that selenium decreased both the incidence of postpartum thyroiditis and permanent hypothyroidism. Selenium administration was also associated with a significant decrease in postpartum TPOAb titers. Given that studies demonstrating benefit are limited, there is insufficient evidence at this time to recommend selenium supplementation during pregnancy in TPOAb-positive women.
Even in women whose thyroid function returns to normal, the risk of recurrent postpartum thyroiditis in a subsequent pregnancy is approximately 70%. Women with a prior history of postpartum thyroiditis should have annual TSH screening to evaluate for permanent hypothyroidism.
Structural Thyroid Disorders in Pregnancy
Goiter
Goiter refers to enlargement of the entire thyroid gland. Goiters can be classified into several categories according to the functional status of the gland (hypothyroid, hyperthyroid, or euthyroid) or to its clinical or scintigraphic appearance (diffuse or multinodular). The most common causes of goiter are summarized in Box 27.9 .
- •
Endemic goiter (iodine deficiency)
- •
Sporadic goiter (diffuse nontoxic goiter; multinodular goiter)
- •
Diffuse toxic goiter (Graves disease)
- •
Thyroiditis (chronic autoimmune [Hashimoto disease]; subacute; silent/postpartum; suppurative)
- •
Drugs (thionamides, iodides, lithium)
- •
Dietary/environmental goitrogens
- •
Organification enzyme defects
- •
Diffuse malignant disease (lymphoma, anaplastic carcinoma)
- •
Infiltrative diseases (Riedel thyroiditis, sarcoidosis, amyloidosis)
Treatment is rarely necessary for diffuse goiter if the patient is asymptomatic and thyroid function testing is normal. With time, however, diffuse enlargement of the thyroid gland typically evolves into multinodular goiter, with progressive autonomous functioning of one or more follicles and occasional progression to thyrotoxicosis. This progression is seen most often in the largest goiters (those with nodules >2.5 cm in size) and in older patients. Large, dominant nodules should undergo fine-needle aspiration, because malignant neoplasms can coexist with this typically benign condition. A trial of thyroid hormone suppression is reasonable in most patients, although less than 50% of nodules will respond to medical therapy by decreasing in size. The approach to therapy in the patient with a toxic multinodular goiter includes thyroid ablation with radioactive iodine, thionamide, or thyroidectomy.
Thyroid Nodules
Nodules of the thyroid are common; they are palpable in 5% of the general population and may be even more common in areas of relative iodine deficiency. Careful attention must be taken to examine the thyroid nodule and surrounding tissues because of the small but real potential of malignancy. Malignant transformation is more common in the largest nodules, in those with progressive growth, in older women, and in women with other risk factors for malignancy (such as prior neck irradiation).
The majority of thyroid nodules are found on pathologic examination to be either hyperplastic or adenomatous in origin. Benign and malignant neoplasms of the thyroid are listed in Box 27.10 . Papillary and follicular carcinomas represent the majority of the cancers. The incidence of thyroid cancer in pregnancy is 1 per 1000. Pregnancy itself does not appear to increase the risk of malignant transformation or alter the course of thyroid cancer. Moreover, treatment for thyroid cancer does not appear to increase the risk of congenital anomalies, low birth weight, or stillbirths.
Benign
- •
Follicular adenoma
- •
Colloid nodule
- •
Hürthle cell adenoma
- •
Multinodular goiter
- •
Simple cyst
- •
Nodular autoimmune thyroiditis
- •
Marine-Lenhart nodule (in Graves disease)
Malignant
- •
Papillary carcinoma
- •
Follicular carcinoma
- •
Hiirthle cell carcinoma
- •
Medullary carcinoma
- •
Anaplastic carcinoma
- •
Lymphoma
- •
Metastases to the thyroid
Any thyroid nodule discovered during pregnancy should be further evaluated, because malignancy may be found in up to 40% of these nodules. Fine-needle aspiration coupled with careful cytopathologic examination of the aspirate is the technique of choice for evaluation of a thyroid nodule. Ultrasound examination may be helpful in distinguishing simple cysts from solitary nodules, in the evaluation of a multinodular goiter, or in the follow-up of known thyroid lesions. However, ultrasonography is a purely anatomic study and does not provide any functional or histologic information. Similarly, scintigraphy (with either technetium or radioiodine) can provide functional information that may be important, because functional (“hot”) nodules are rarely malignant, and almost all carcinomas are nonfunctional (“cold”). However, such testing cannot definitively exclude malignancy. As such, neither of these diagnostic tests can replace fine-needle aspiration for the initial evaluation of a thyroid nodule.
If a diagnosis of thyroid cancer is made, a multidisciplinary treatment plan should be established. Management options include pregnancy termination, treatment during pregnancy, and preterm or term delivery with definitive treatment after pregnancy. The decision will be affected by the gestational age at diagnosis and by the tumor characteristics. Definitive treatment for thyroid cancer is thyroidectomy and radiation. If necessary, thyroidectomy can be performed during pregnancy, preferably in the second trimester. However, given the slow progression of most thyroid cancers, surgery can often be delayed until after delivery. Radiation is best deferred until after pregnancy.
Disorders of Calcium Metabolism
- ◆
Pregnancy is associated with a net accumulation of calcium, primarily due to elevated circulating levels of biologically active 1,25-hydroxyvitamin D (cholecalciferol), leading to an increase in calcium absorption from the gastrointestinal tract. Changing levels of parathyroid hormone (PTH) do not appear to be a driver of the accumulation of calcium that occurs in pregnancy. Calcitonin levels do not change in pregnancy.
- ◆
Calcium is actively transported across the placenta against a concentration gradient to the fetal compartment. The fetoplacental unit sequesters calcium to build the fetal skeleton.
- ◆
Compared with the mother, the human fetus is relatively hypercalcemic, hypercalcitonemic, and hypoparathyroid.
Total calcium stores in the mother are distributed between a large skeletal pool of “inert” calcium (1 kg) and a small extracellular pool of bioavailable calcium. These two pools of calcium are maintained in a state of dynamic equilibrium, controlled on the one hand by PTH and by calcitonin on the other. PTH stimulates release of calcium from bone and promotes calcium uptake from the gastrointestinal tract, while calcitonin suppresses calcium release from bone. Calcium uptake from the gastrointestinal tract is also regulated by vitamin D metabolites. Calcium is excreted by the kidneys and is sequestered by the fetoplacental unit to build the fetal skeleton.
Pregnancy is associated with a net accumulation of calcium. At term, the total accumulation of calcium in the mother is approximately 25 to 30 g, most of which is sequestered in the fetal skeleton. This is due primarily to elevated circulating levels of biologically active 1,25-hydroxyvitamin D (cholecalciferol), leading to an increase in calcium absorption from the gastrointestinal tract. The decidua may be a major source of 1,25-hydroxyvitamin D in pregnancy. Calcitonin levels do not change in pregnancy. Although initial studies suggested that serum PTH levels increase during pregnancy, subsequent studies using more sensitive dual-antibody assays have shown that PTH levels are lower throughout gestation. Urinary excretion of calcium increases during pregnancy, but the ratio of urinary calcium to creatinine decreases, suggesting an attempt by the kidneys to reabsorb and conserve calcium, even in the face of an increased glomerular filtration rate. In general, bone density remains relatively constant during pregnancy, although some investigators have reported a slight decrease in bone density in the third trimester. Pregnancy is also associated with a decrease in serum albumin and a concomitant decrease in total calcium concentrations ( Fig. 27.16 ). The upper limit of normal for total serum calcium in pregnancy is approximately 9.5 mg/dL. However, ionized calcium concentrations do not change significantly during pregnancy.
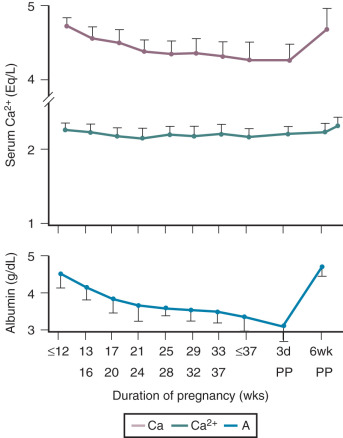
Calcium from the maternal compartment is actively transported across the placenta against a concentration gradient to the fetal compartment. This process is regulated, at least in part, by the production of PTH-related protein (PTHrP), a PTH homologue, by the fetal parathyroid glands. PTHrP levels rise in maternal serum throughout gestation. In mice lacking the gene coding for PTHrP, calcium transport from the maternal to the fetal compartment is markedly impaired but can be rescued completely by administration of exogenous PTHrP. Compared with the mother, the human fetus is relatively hypercalcemic, hypercalcitonemic, and hypoparathyroid. Separation of the fetus from the mother is associated with a fall in serum calcium and a compensatory rise in serum PTH and a fall in calcitonin levels.
Hyperparathyroidism
Primary hyperparathyroidism in pregnancy is rare; only a few hundred cases have been reported. Causes of hyperparathyroidism include a solitary parathyroid adenoma (80% of cases), generalized hyperplasia (15%), multiple adenomas (3%), and carcinoma (<2%). Maternal complications of untreated or poorly controlled hyperparathyroidism include hyperemesis gravidarum, generalized weakness, headache, confusion, emotional lability, nephrolithiasis, pancreatitis, and hypertension. Spontaneous abortion and perinatal mortality rates are also increased in pregnancies complicated by hyperparathyroidism ; however, improved diagnosis and treatment have led to a substantial decrease in perinatal mortality. Most authorities recommend surgical excision of the parathyroid adenoma in symptomatic women, but controversy persists as to the optimal management for asymptomatic women and women with mild hyperparathyroidism. At birth, neonatal hypocalcemic tetany is common and typically occurs in the first 2 weeks of life.
Unusual causes of hypercalcemia in pregnancy include familial hypocalciuric hypercalcemia (FHH) and sporadic cases of inappropriate secretion of PTHrP. Women with FHH typically present with mild hypercalcemia, mild elevations in circulating PTH concentrations, and low urinary calcium. Because of the autosomal-dominant nature of the disease and high penetrance, infants can present with either hypercalcemia (if the neonate has FHH) or hypocalcemia (if the neonate does not have FHH but is responding to maternal hypercalcemia).
Hypoparathyroidism
The most common cause of maternal hypoparathyroidism is incidental resection of the parathyroid glands at the time of thyroidectomy. This complication occurs in approximately 1% of thyroidectomy cases. Symptoms of hypocalcemia include numbness and tingling of the fingers and orofacial area. Chvostek sign (twitching of the facial muscles when the facial nerve is tapped) and Trousseau sign (induction of carpopedal spasm by applying pressure to the upper arm with a blood pressure cuff) are often present.
If untreated, maternal hypocalcemia can lead to compensatory hyperparathyroidism in the fetus, leading to bone demineralization. The treatment of maternal hypoparathyroidism is calcium (1.2 g daily) and either vitamin D (50,000 to 150,000 IU daily) or the active metabolite, calcitriol (0.25 to 3 µg daily). If circulating calcium levels can be maintained at or near the normal range, pregnancy outcome will not be adversely affected. In women with hypocalcemia, labor may be complicated by generalized tetany that requires intravenous calcium administration. Vitamin D is secreted into breast milk and may lead to hypercalcemia in the newborn. As such, breastfeeding may not be advisable in women receiving high-dose vitamin D therapy.
Related posts:
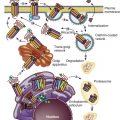 Gonadotropin Hormones and Their Receptors
Gonadotropin Hormones and Their Receptors
 Steroid Hormones and Other Lipid Molecules Involved in Human Reproduction
Steroid Hormones and Other Lipid Molecules Involved in Human Reproduction
 Structure, Function, and Evaluation of the Female Reproductive Tract
Structure, Function, and Evaluation of the Female Reproductive Tract
 Endocrine Disturbances Affecting Reproduction
Endocrine Disturbances Affecting Reproduction
 Physiologic and Pathophysiologic Alterations of the Neuroendocrine Components of the Reproductive Axis
Physiologic and Pathophysiologic Alterations of the Neuroendocrine Components of the Reproductive Axis
 Pelvic Imaging in Reproductive Endocrinology
Pelvic Imaging in Reproductive Endocrinology
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


