Endocrine and genetic bases of hormone-related cancers
Leslie Bernstein, PhD  Xia Pu, PhD
Xia Pu, PhD  Jian Gu, PhD
Jian Gu, PhD
Overview
Neoplasia of hormone-responsive tissues currently accounts for more than 30% of all newly diagnosed male cancers and almost 40% of all newly diagnosed female cancers in the United States. Given that endogenous hormones apparently affect the risk of these cancers and their overall frequency, concern exists about the effects on cancer risk if the same or closely related hormones are administered for therapeutic purposes (e.g., as contraceptives, as menopausal hormone therapy, or for the prevention of miscarriage). Depending on the timing of hormone use and the tissue-specific effects, some of these compounds will reduce risk while others increase risk of “hormone-dependent” cancers. This chapter focuses on breast, endometrial, and ovarian cancers among women and prostate cancer among men and provides a review of the epidemiologic and endocrinologic evidence for the role of hormones in cancer development. It also reviews the current status of the relationship between exogenous hormones and risk of cancers of the breast, endometrium, and ovary. In addition, it summarizes our current knowledge of genetic susceptibility to breast, endometrial, ovarian, and prostate cancers. Other less common cancers (e.g., cervical cancer, clear cell vaginal adenocarcinoma, thyroid cancer, testicular cancer, and osteosarcoma) may have a hormonal basis as well, but are not reviewed in this chapter.
Substantial and convincing bodies of experimental, clinical, and epidemiologic evidence indicate that hormones play a major role in the etiology of several human cancers. The concept that hormones can increase the incidence of neoplasia was first proposed by Bittner,1 on the basis of experimental studies of estrogens and mammary cancer in mice. This theory has been refined into epidemiologic hypotheses related to cancers of the breast, endometrium, prostate, ovary, thyroid, bone, and testis.2, 3 The underlying mechanism proposed for these cancers is that neoplasia is the consequence of prolonged hormonal stimulation of the target organ, the normal growth and function of which is controlled by one or more steroid or polypeptide hormones. Evidence is mounting to show that the amount of hormone to which a tissue is effectively exposed is under strong genetic control.4 Therefore, although external factors such as physical activity or exogenous hormone use may modify hormone profiles, the current evidence supports a multigenic model of cancer susceptibility.5, 6
The major carcinogenic consequence of this hormonal exposure at the end organ is cellular proliferation, although direct carcinogenesis resulting from metabolic activation and direct DNA (deoxyribonucleic acid) binding is another potential mechanism. The emergence of a malignant phenotype depends on a series of somatic mutations that occur during cell division, but the entire sequence of genes involved in progression from normal cell to a particular malignant phenotype is not known (Figure 1). Candidate genes include those in the endocrine and growth factor pathways,4, 7, 8 as well as DNA repair genes, tumor suppressor genes, and oncogenes.9, 10 Germline mutations have been described in two such tumor suppressor genes, BRCA1 and BRCA2, which are associated with susceptibility to breast and ovarian cancer.11–14 Germline mutations in TP53 are also associated in certain kindreds with an increased risk of breast cancer.15 However, mutations in these genes do not appear to be involved in the majority of sporadic breast cancer. Recent genome-wide association studies (GWAS) have identified many common, low-penetrance susceptibility loci for sporadic cancers.16
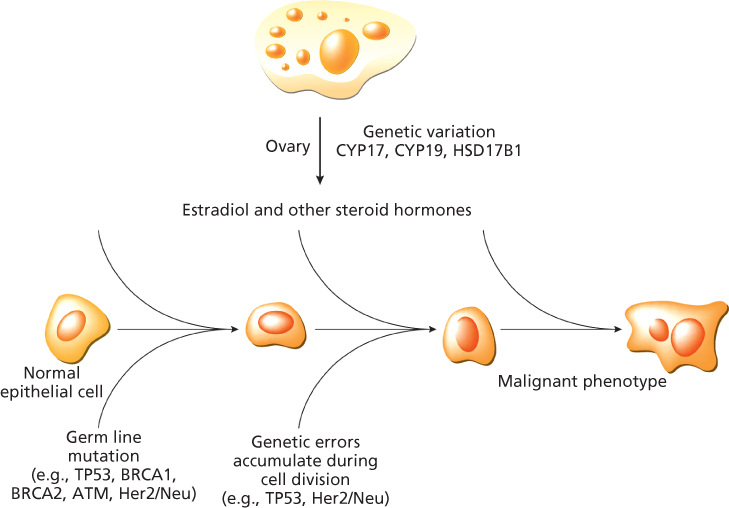
Figure 1 Estradiol and other steroid hormones (e.g., progesterone) drive breast cell proliferation, which facilitates the accumulation of random DNA copying errors in critical genes on the pathway to a malignant phenotype. Germ line mutations in relevant tumor suppressor genes accelerate the transformation to the malignant phenotype.
Neoplasia of hormone-responsive tissues currently accounts for more than 30% of all newly diagnosed male cancers and almost 40% of all newly diagnosed female cancers in the United States. Given that endogenous hormones apparently affect the risk of these cancers and their overall frequency, concern exists about the effects on cancer risk if the same or closely related hormones are administered for therapeutic purposes (e.g., as contraceptives, as menopausal hormone therapy, or for the prevention of miscarriage).17 This chapter focuses on breast, endometrial, and ovarian cancers among women and prostate cancer among men and provides a review of the epidemiologic and endocrinologic evidence for the role of hormones in cancer development. It also reviews the current status of the relationship between exogenous hormones and risk of cancers of the breast, endometrium, and ovary. Other less common cancers (e.g., cervical cancer, clear cell vaginal adenocarcinoma, thyroid cancer, testicular cancer, and osteosarcoma) appear to have a hormonal basis as well, but are not reviewed here.
Breast cancer
Breast cancer is the most common cancer in women; it is expected that 232,670 new cases of invasive breast cancer and 62,570 new cases of breast carcinoma in situ will be diagnosed in the United States in 2014 and that 40,430 United States women will die of breast cancer.18 A perceptible decline in breast cancer mortality has occurred since 1990,19 resulting from greater use of mammographic screening, hormonal therapy, and therapy that targets HER2/neu.20, 21 Available evidence regarding the hormonal etiology of breast cancer is most consistent with the hypothesis that estrogen is the primary stimulant for breast cell proliferation.2, 3 The simultaneous presence of progesterone further increases the rate of proliferation.22 This latter conclusion is based largely on the fact that breast mitotic activity peaks during the luteal phase of the menstrual cycle23 and it is consistent with the increasing number of publications demonstrating that added progestins substantially augment the increased risk of breast cancer from estrogen therapy (ERT).24–28
The most consistently documented, hormonally related risk factors for breast cancer are early age at menarche, late age at menopause, late age at first full-term pregnancy, and excess weight (Table 1). The age-incidence curve for breast cancer emphasizes the importance of ovulation in determining risk.17 Cases first occur during early adulthood, and the rate of increase in incidence then rises sharply with age until the time of menopause, when it slows dramatically. The rate of increase in the postmenopausal period is approximately one-sixth the rate of increase in the premenopausal period. This age-incidence curve appears, then, to be shaped in a major way by the effects of ovarian activity. Therefore, it is critical to understand the determinants, both genetic and environmental, of the onset, regularity, and cessation of ovulation in order to continue to develop effective prevention modalities for breast cancer.
Table 1 A summary of established hormonal risk and protective factors for breast cancer
|
|
Reproductive factors
Early age at menarche is an established risk factor for breast cancer.3 In general, risk decreases approximately 5–6% for each year that menarche is delayed, and this relationship may be further modified by the age at onset of regular ovulatory menstrual cycles. In a study of young women, Henderson et al.29 reported that women with menarche at age 12 years or younger who experienced rapid onset of regular cycles had nearly a fourfold greater breast cancer risk than women with later menarche who experienced a long duration of irregular cycles. This is further supported by results from a study that showed that circulating estrogen and progesterone levels in daughters of women with breast cancer were higher than those of age-matched daughters of women without breast cancer.30
Although menarche and the onset of ovulation are to some extent genetically determined,31 it is also critical to establish behaviors that may alter the number of ovulatory menstrual cycles a woman experiences during her reproductive years. Strenuous physical activity may delay menarche.32 For example, in one study, the mean age at menarche of ballet dancers was 15.4 years compared to 12.5 years for control subjects. Moderate physical activity during adolescence can lead to anovulatory menstrual cycles. Girls who regularly engaged in moderate levels of physical activity (averaging at least 600 kcal of energy expended per week during a 9-month school year) were nearly three times more likely to have anovulatory menstrual cycles than were girls who were less physically active.33 Bernstein et al.34 have reported that lifetime patterns of leisure-time exercise activity significantly impact the breast cancer risk in young women (<40 years of age), older, postmenopausal women (55–64 years of age),35 and African-American and Asian-American women.36, 37 Evidence continues to accumulate in support of a protective effect of physical activity on breast cancer risk with risk reductions observed in both case-control and cohort studies, although studies may vary according to the subgroups of women who benefit the most.38–40
Later occurrence of menopause and extended exposure to ovulatory cycles at the end of menstrual life also increase breast cancer risk. The risk of women whose natural menopause occurs before age 45 years is one-half that of women whose menopause occurs after age 55 years.41 Artificial menopause, induced by bilateral oophorectomy or by pelvic irradiation, also markedly reduces breast cancer risk; this reduction is greater than that associated with natural menopause before age 50 years.41–43 Following natural menopause, estrogen exposure declines gradually due to the continuing, but declining function of the ovaries and the continuing, persistent ovarian production of a small amount of testosterone.
The relationship between weight and breast cancer risk depends on menopausal status. Among postmenopausal women, a 10-kg increment in body weight results in about an 80% increase in breast cancer risk.44 One explanation for this effect is that heavier postmenopausal women have higher circulating estrogen levels because of the conversion of an adrenal androgen, androstenedione, to estrone by aromatase present in body fat. In premenopausal women, the relationship between weight and risk is less clearly established, but if anything, the situation is the reverse of that in postmenopausal women; here, high weight is associated with reduced risk.45 This may result from a reduction in the frequency of ovulatory menstrual cycles associated with high body weight.
Assuming ovarian activity affects breast cancer risk, case-control and cohort studies of breast cancer should find higher levels of circulating estradiol among breast cancer patients than among healthy women. Bernstein et al.46 described the results of two concurrent case-control studies of premenopausal women in the United States (Los Angeles) and China (Shanghai). Overall, breast cancer patients had 14% higher serum estradiol concentrations, with a case-to-control excess of 17% in Chinese women and 11% in white American women, respectively. Los Angeles control women had 21% greater estradiol concentrations than did Shanghai control women, and adjustment for body weight only accounted for 25% of this difference. The results from a pooled analysis of nine prospective studies of endogenous hormones and postmenopausal breast cancer risk provide strong evidence that high estradiol concentrations are predictive of breast cancer risk, with women in the highest quintile of estradiol exposure having a twofold greater breast cancer risk than those in the lowest exposure quintile.47
Age at first birth
Having a first full-term pregnancy at a young age (i.e., before age 20 years) lowers a woman’s breast cancer risk by about 50% relative to nulliparous women. Full-term pregnancies at later ages provide smaller increments of protection.48 Women who have a late first full-term pregnancy (i.e., in their thirties) have greater breast cancer risk than nulliparous women have. This paradoxical cross-over effect of a late first full-term pregnancy has been repeatedly confirmed by epidemiologic studies.
The immediate effect of a full-term pregnancy on breast cancer risk is a short-term increase. Among women who have given birth within the past 3 years, breast cancer risk is nearly three times higher than that of women of the same age, parity, and age at first birth whose most recent birth occurred at least 10 years earlier.49 On the basis of these results, it appears that a first pregnancy confers two contradictory effects on breast cancer risk: a short-term increase in risk, followed in the long term by a substantial reduction in risk.49
This apparent paradox has a physiologic explanation based on patterns of estrogen as well as prolactin secretion and metabolism during pregnancy. During the first trimester, the level of bioavailable estradiol rapidly rises, an effect that is more apparent during the first than in subsequent pregnancies.50 Thus, in terms of estrogen exposure to the breast, the net effect during this early part of pregnancy is an increased risk that is equivalent to the exposure from several ovulatory cycles over a relatively short period of time.51 At a molecular level, it is likely that the hormonal changes during pregnancy induce irreversible differentiation and apoptosis in some cells that have already accumulated one or more of the relevant somatic mutations necessary for breast cancer development. In the long run, however, this negative effect of early pregnancy on breast cancer risk can be overridden by two beneficial hormonal consequences of completing the pregnancy. It has been reported that prolactin levels are substantially lower in parous compared to nulliparous women.52–54 Prolactin, a polypeptide hormone, regulates lactation and appears to enhance estrogen effects on breast tissue. In addition, parous women have been reported to have lower levels of bioavailable estradiol than nulliparous women.51
Evidence is fairly convincing that lactation reduces the breast cancer risk of premenopausal women,55–57 but is less consistent for postmenopausal women.56, 58 In two publications, Enger et al.55, 58 showed substantial breast cancer risk reductions, of 35% for premenopausal and 30% for postmenopausal parous women in the United States who breast fed for more than 15 months (relative to similar women who never lactated). In the United States, rates of breast feeding have varied over time; some studies may have not observed lower breast cancer risk among women who have breast fed due to the small proportion of women with a sufficient duration of lactation. Among premenopausal and postmenopausal women in Shanghai, where breast feeding often extends to more than 1 year per child, a strong dose–response effect of decreasing breast cancer risk with increasing breast feeding duration was observed.59 The time when supplementary feedings are introduced to the infant as well as the frequency and duration of each breast feeding episode may also contribute to the inconsistent findings. Lactation may reduce breast cancer risk by reducing the total number of ovulatory menstrual cycles a woman experiences because breast feeding results in a substantial delay in reestablishing ovulation following a completed pregnancy.
Diet
Much attention has focused on dietary differences between countries, particularly fat consumption patterns, to explain both the international pattern of breast cancer occurrence and changes in rates of breast cancer following migration to high-risk from low-risk countries.60 International breast cancer mortality and incidence rates correlate highly with per capita consumption of fat in the diet (correlation coefficient, r = 0.93 and r = 0.84, respectively).60 As implied previously, nutrition may influence breast cancer occurrence by modifying age at menarche and body weight, but the correlation of fat consumption with international breast cancer mortality remains highly significant, even after statistical adjustment for these factors.
Although it has been theorized that fat intake in the diet may be an important contributor to breast cancer risk, many case-control studies of fat consumption and breast cancer find only small differences between cases and controls. Similarly, most of the cohort studies that have used food-frequency questionnaires to study the relationship with total fat, saturated fat, or vegetable fat have found little or no difference in breast cancer risk over a wide range of fat intake. In a meta-analysis of studies of total fat intake and breast cancer risk, the extent of increase in risk comparing women in the highest intake category with those in the lowest intake category was 14% for case-control studies (summary odds ratio (OR) =1.14; 95% confidence interval (CI) = 0.99–1.32) and 11% for cohort studies (OR = 1.11; 95% CI = 0.99–1.25).61 The Women’s Health Initiative randomized trial of more than 48,000 women tested the hypothesis that reducing intake of total fat to 20% of energy and increasing consumption of vegetables and fruit to at least five servings daily and grains to at least six servings daily would lower cancer, and particularly breast cancer, risk.62 During 8.1 years average follow-up after randomization, the RR (relative risk) was lower in women who adhered to the diet compared to the nondietary intervention group, although the results were not quite statistically significant (RR = 0.91; 95% CI = 0.83–1.01), but were suggestive of an impact of diet on breast cancer risk.
High-fiber diets may protect against breast cancer, perhaps because fiber reduces the intestinal reabsorption of estrogens excreted via the biliary system.63 Assessment of fiber intake in epidemiologic studies has been problematic because of a paucity of data on the fiber content of individual foods and disagreement about the most appropriate methods of biochemical analysis to determine the different types of fiber. Case-control, but not cohort, studies have shown a consistent inverse association between dietary fiber intake and breast cancer risk.64
Several investigations have been undertaken to demonstrate a reduction in serum estrogen levels following dietary interventions that reduce fat or increase fiber intake.65, 66 A meta-analysis of these studies demonstrated a 7.4% average reduction in estradiol levels of premenopausal women and a 23% reduction in estradiol in postmenopausal women following trials of reduced dietary fat intake.66 This analysis could not distinguish between a direct dietary effect on hormone levels and an indirect effect that was due to disruption of ovulatory cycles in premenopausal women; nevertheless, whatever the mechanism, such a reduction in estradiol levels is potentially very important.
Exogenous hormones
Hormone therapy and oral contraceptives are the exogenous counterparts to endogenous hormonal exposures experienced by women and therefore are of concern as potential contributors to breast cancer risk.
Oral contraceptives
The relationship between oral contraceptive use and breast cancer risk has been the topic of many review articles. A combined analysis of 54 studies that included more than 150,000 women has provided many important answers about the risk of breast cancer among users of combination oral contraceptives (COCs), which combine an estrogen and a progestin in a single pill.67 This analysis indicates a modest increased risk of breast cancer among current (RR = 1.24) and recent (RR = 1.16) COC users. Age at first COC use modifies the association with recent use. For recent users, the risks are highest for those who began COC use before the age of 20 years. However, total duration of COCs use was not associated with increased risk of breast cancer, once recency of use was taken into account.
The pooled analysis compiled studies that mainly focused on younger women as most of these studies were conducted at a time when oral contraceptive users had not achieved their perimenopausal and postmenopausal years.67 The Women’s Contraceptive and Reproductive Experiences (Women’s CARE) Study, a population-based case-control study conducted in five geographic regions of the United States,68 involved more than 4500 newly diagnosed breast cancer patients and more than 4500 control subjects, all of whom were 35–64 years of age. This study was specifically designed to assess the impact of oral contraceptives among women who no longer used oral contraceptives. Relatively few participants in the Women’s CARE Study were current oral contraceptive users; many women had stopped use at least 20 years earlier. No significant associations were observed between duration of use, estrogen dose of the formulation, age at first use, interval since last use, or use in relation to timing of pregnancy and breast cancer risk. Further, results for younger women (35–44 years) were similar to those for older women (45–64 years) who were more likely to have used earlier formulations, but were less likely to have recently used oral contraceptives.
The International Agency for Research on Cancer completed a review of all of the existing literature on COCs and breast cancer risk concluding that breast cancer risk is increased in current or recent oral contraceptive users, particularly among women under age 35 years whose first oral contraceptive use was before age 20 years.69 The increase in risk disappeared as age at current or recent use increased and, following cessation of oral contraceptive use, any increase in risk disappeared within 10 years.
Hormone therapy
Hormone therapy has evolved over time. Originally designed to reduce the symptoms of menopause, hormone therapy gained popularity because of its efficacy in reducing the risk of osteoporosis and its purported benefits in reducing the risk of heart disease. Initially formulated as ERT, the number of women using hormone therapy increased through the mid-1970s, until concerns were raised about the carcinogenic potential of ERT on the endometrium. In the 1980s, cyclic estrogen–progestin regimens became widely recommended and prescribed to eliminate the increase in endometrial cancer associated with estrogen-alone therapy. Initially, these were prescribed as sequential regimens with estrogen given during the first 15–20 days of a 28-day cycle followed by 5–10 days when both estrogen and progestin were given. More recently, continuous combined regimens have gained favor because they reduce menstrual-like bleeding episodes and because of their ease of administration.
Most studies that have included sufficiently large numbers of women who have used ERT for extended periods of time (e.g., for more than 10 years) indicate a modest increase in breast cancer risk among exposed women, with risk increasing approximately 3% per year of use.70 Among studies conducted in the United States, where the use of conjugated equine estrogens is the norm, it was estimated that breast cancer risk increased about 2.2% per year of use of a standard dose regimen (0.625 mg/day).
The Collaborative Group on Hormonal Factors in Breast Cancer pooled data from 51 epidemiologic studies and more than 160,000 women to assess the impact of hormone therapy on breast cancer risk.55 Where information was available regarding type of hormone preparation used, 80% of women in these studies had used mostly estrogen-only therapy and 12% of women had used combination hormone therapy. This study showed that hormone therapy (primarily ERT) increased breast cancer risk with RRs of 1.09, 1.15, and 1.14 comparing ever users to never-users in cohort studies, population-based case-control studies, and hospital-based case-control studies, respectively. Risk was substantially elevated among women who had used hormone therapy for at least 15 years (RR = 1.58). Risk increased by 2.3% (p = 0.0002) for each year of use among women who had used hormone therapy within 5 years of diagnosis or an equivalent reference date (Figure 2). However, women who stopped hormone therapy use 5 or more years earlier had only a modest, nonsignificant increase in risk, regardless of duration of use.
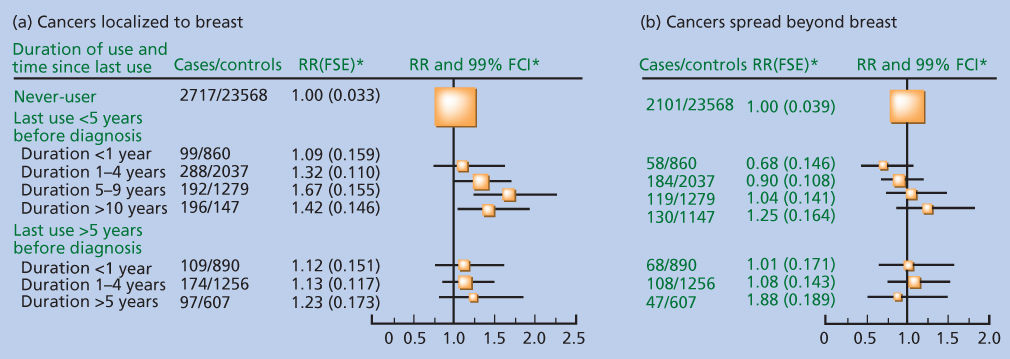
Figure 2 (a,b) Relative risk of breast cancer by duration and time since last use of hormone therapy according to extent of tumor spread relative to never-users, stratified by study, age at diagnosis, time since menopause, body mass index, parity, and the age of a woman when her first child was born. “Last use within 5 years before diagnosis” includes current users. *Floated standard errors (FTEs) and floated confidence intervals (FCIs) calculated from floated variance for each category.
Consistent with these estimates are the results from the Million Women Study, conducted in the United Kingdom, which recruited a cohort of women aged 50–64 years who had undergone routine mammography.61 In this study, incidence of breast cancer was significantly greater among current users of ERT than among women who had never used hormones (RR = 1.30; 95% CI = 1.22–1.38). Risk increased with increasing duration of use among these current users, with 5–9 years of use associated with a RR of 1.32 and 10 or more years of use associated with a RR of 1.37.
The Women’s Health Initiative randomized trial compared an estrogen-alone regimen to placebo among women 50–79 years of age who had previously had a hysterectomy and assessed the impact of a combined estrogen plus progestin regimen versus placebo among similarly aged women who had an intact uterus. The estrogen-alone regimen consisted of 0.625 mg/day of conjugated equine estrogen,71 and the combined regimen consisted of 0.625 mg/day of conjugated equine estrogen and 2.5 mg/day of medroxyprogesterone acetate.27 The results for the ERT study were somewhat surprising in that, after an average follow-up of 7.1 years, the RR of breast cancer was not elevated (RR = 0.80; 95% CI = 0.62–1.04).72 In the trial, the reduction in risk was greater and was statistically significant for ductal cancers although even in a trial this large, numbers were too small to demonstrate a difference in risk by tumor histology. In comparison to ductal tumors, risk appeared elevated for lobular cancers and the comparison of ductal to lobular cancer was of borderline statistical significance (p = 0.054). A similar, nonstatistically significant difference was observed by stage with risk reduced for localized cancers but not for regional cancers (p = 0.09).
For combined hormone therapy (CHT), the Women’s Health Initiative assessed only the continuous combined regimen. Three population-based observational studies, published in 1999 and 2000, showed that CHT conferred a greater risk of breast cancer than did an estrogen-alone regimen.24, 25, 73 For example, Ross et al.25 found that the increment in risk for each 5 years of use was nearly four times greater for CHT users than for ERT users. Results from the Women’s Health Initiative trial arm comparing a continuous CHT to placebo provided a risk estimate that was similar to those from these prior studies (RR = 1.24; 95% CI = 1.01–1.54).27
Lee et al.74 conducted a meta-analysis of the results of studies that have evaluated the impact of CHT on breast cancer risk, separating results for use of sequential (cyclic) combined regimens from those for use of continuous combined regimens and including results from the Collaborative Group on Hormonal Factors in Breast Cancer55 and the Women’s Health Initiative.27 Overall, users of a combined regimen had substantially elevated risk that increased 7.6% per year of use.74 Not all studies provided data on progestin schedule. Those that did showed a small difference in risk between sequential and continuous combined regimens (increase in breast cancer risk per year of use of 8.9% and 10.3%, respectively). Notably, results from Scandinavian studies showed that continuous regimens had a greater impact on risk than sequential regimens. This difference was not as apparent among studies from the United States or the Million Women’s Study. In the United States, the total dose of progestin is comparable in continuous combined compared to sequential regimens, whereas in Scandinavia, continuous combined regimens provide a substantially higher dose of progestin. Thus, evidence is quite strong that the progestin component of combined regimens adds substantially to any increase in breast cancer risk conferred by estrogen alone and that differences in results between the United States and Scandinavia are likely due to differences in the progestin dose administered.
Recently, the Women’s Health Initiative investigators published an update on health risks and benefits of CHT, examining risk following cessation of hormone therapy.75 Risk for invasive breast cancer remained elevated after an average 2.4-years follow-up after use was stopped among women who had been randomized to the estrogen plus progestin arm of the trial evaluating combined therapy (RR = 1.27; 95% CI = 0.91–1.78), although the CI did not exclude 1.0.75
CHT preferentially increases the risk of lobular and ductal-lobular breast cancers, particularly those judged to have more than a 50% lobular component.76 Among older women, ages 55–74 years, current users of combined therapy were at 2.7-fold greater risk of lobular carcinoma and 3.3-fold greater risk of ductal-lobular carcinoma compared to women who had no use of combined therapy.
Declining use of combined therapy has had a marked impact on breast cancer incidence rates in the United States and Germany.77–79 It is important to note that rates of breast cancer in the United States began to decline before publication of the Women’s Health Initiative result for combined therapy in 2002.26 An evaluation of data from the Surveillance, Epidemiology, and End Results (SEER) registries for 1975–2003 shows a decline in invasive breast cancer from 1999 onward in all age groups of women 45 years or older, with a sharp decrease in incidence in 2002 and 2003, particularly of estrogen receptor- (ER) positive tumors, in women 59–69 years of age.78 Data from a mammography registry in San Francisco indicate that hormone therapy prescribing peaked in 1999, then began to decline, which was amplified following publication of the Women’s Health Initiative.80 Robbins and Clarke81 confirmed this in their analysis of breast cancer incidence across 58 counties in California, clearly demonstrating that breast cancer rates declined between 2001 and 2004, with the rate of decline paralleling the reduction in prescriptions of combined therapy recorded by the California Health Interview Survey. Although some have suggested that part of this decline in breast cancer incidence among older women might be due to decreasing use of mammographic screening, rates of in situ breast cancer, which is diagnosed almost exclusively by mammography, have not decreased in parallel with the decrease in invasive breast cancer,78 and the California Health Interview Survey has not indicated any significant change in the proportion of women reporting a screening mammogram within the prior 2 years.81
Endometrial cancer
Among the hormone-related cancers, etiologically the best understood is endometrial cancer. All the major demographic characteristics of the disease, as well as the major nondemographic risk factors, are explicable on the basis of cumulative exposure of the endometrium to that fraction of estrogen, which is unopposed by the modifying influences of progesterone.2, 3
Mitotic activity in the endometrium
Key and Pike82 summarized the existing data on endometrial mitotic activity during normal menstrual cycles. Mitotic rates are low during days 1–4 of the cycle, then increase rapidly and remain stable thereafter until day 19, after which rates essentially drop to zero for the remainder of the cycle. There appears to be a lag period of about 4 days before the stimulatory effects of unopposed estrogen or the modifying influence of progesterone on endometrial mitotic activity are fully apparent.
The cellular basis for the antiestrogenic activity of progestogens on the endometrium is well understood.2 Progestogens reduce the concentration of estradiol receptors and increase the activity of the 17-β-hydroxysteroid dehydrogenase type II enzyme, which converts estradiol to estrone,83, 84 a biologically less-potent estrogen because of its lower affinity for cellular ERs. Luteal phase progesterone causes endometrial cells to differentiate to a secretory state and progestogen withdrawal leads to cyclic sloughing of endometrial tissue.
On the basis of the concept that frequency of mitotic activity is the primary determinant of endometrial cancer risk and that such activity is controlled by cumulative exposure to unopposed estrogens, one can readily predict the most important risk factors for this disease (Table 2). Pregnancies and oral contraceptives, which expose the endometrium to constant high levels of both estrogen and progestogen, should protect against endometrial cancer development. ERT and obesity should increase the risk. All of these predicted effects have been repeatedly well documented in epidemiologic studies.2
Table 2 A summary of established hormonal risk and protective factors for endometrial cancer
|
|
Estrogen therapy
Hormone therapy in the form of unopposed ERT gained widespread popularity in the United States during the 1960s and 1970s.85 Concomitantly, incidence rates of endometrial cancer in postmenopausal women also increased rapidly, especially in western US states, where use of ERT was particularly common.86 By 1975, the results of epidemiologic case-control studies, demonstrating a strong overall association between ERT and risk of endometrial cancer, were being published.87, 88 Dozens of studies have now documented a high relative increase in the risk of endometrial cancer following ERT. Risk is strongly related both to dose and to duration of use, but high relative increments in risk follow even moderate doses taken for intermediate length periods of time. Women who use ERT for 5 years or longer have approximately a 3.5-fold increase in risk compared to that of women who have never used such therapy (Figure 3a).17
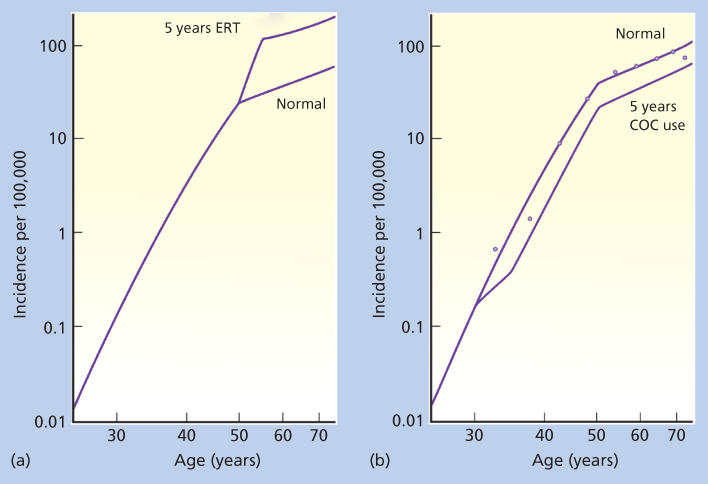
Figure 3 Age-specific incidence rates for cancers of the endometrium in women using estrogen therapy (ERT) (a) and combination oral contraceptives (COCs) (b) for 5 years. Data are from the UK Birmingham Cancer Registry for the years 1968–1972. These data largely avoid problems arising from the high hysterectomy and oophorectomy rates in the United States, which artificially distort the age-incidence curves. Dots, actual incidence data; solid lines marked “normal,” mathematic models predicting these rates from the major known risk factors for these cancers.
Source: Henderson et al. 1993.17 Reproduced with permission from The American Association for the Advancement of Science.
Although use of estrogen clearly increases the incidence of aggressive endometrial cancer, the overall mortality from endometrial cancer among affected users somewhat paradoxically is much lower than among nonusers who develop endometrial cancer.89 In fact, such women have little reduction in life span when compared to healthy women of the same age. The reasons for this are not completely known, but this phenomenon likely can be explained by the increased medical surveillance among estrogen users. Women who use ERT tend to be closely monitored because the drug frequently induces vaginal bleeding. Part of the favorable survival experience may also result from patients with estrogen-induced benign hyperplasia being misdiagnosed as having endometrial cancer. Although past users of ERT have a risk of endometrial cancer that is intermediate between that for current users of comparable duration and lifetime nonusers, risk in such women remains substantially elevated over baseline even after many years without treatment.90
As noted above, the newer regimens of hormone therapy typically follow one of two patterns: sequential or combined estrogen and progestin. The sequential regimen attempts to reproduce the hormonal pattern of the normal menstrual cycle, albeit at lower levels of both estrogen and progestogen. One therefore might predict that this method of hormone therapy administration would only partially offset the increased risk of endometrial cancer that is associated with unopposed ERT. Pike et al.91 showed that if progestins are added for less than 10 days per month, the risk is only slightly reduced. However, regimens that include progestins for more than 10 days in a month, or where progestins are given continuously with estrogen, do not increase endometrial cancer risk.91
The Million Women Study conducted in Great Britain, which included 716,738 postmenopausal women without prior cancer or hysterectomy who were recruited between 1996 and 2001, also provides data on this issue.74 Relative to women who had never used any hormonal therapy, endometrial cancer risk was substantially lower among women who had used continuous CHT as their most recent hormone therapy, with a RR of 0.71 (95% CI = 0.56–0.90). For women using cyclic CHT, however, risk did not differ from that of nonusers (RR = 1.05; 95% CI = 0.92–1.22). The number of cases among women taking estrogen-alone therapy was small, as, since the mid-to-late 1970s, this has rarely been given to women with an intact uterus. Among women on estrogen-alone therapy, the RR of endometrial cancer was substantially lower than prior studies had observed (RR = 1.45; 95% CI = 1.02–2.06). As expected, based on the fact that obesity increases endometrial cancer risk through an estrogen pathway causing endometrial proliferation,82, 92 the reduction in risk on CHT was greatest among obese women and the increase in risk for estrogen-alone therapy was greatest among nonobese women.
The authors of the Million Women Study also provided an extensive assessment of published studies, including their own, on the impact of CHT on endometrial cancer risk.74 Overall, they calculated a summary estimate that the RR for continuous CHT across all studies was 0.88 (95% CI = 0.75–1.03) compared to never-users. This estimate, however, was based on relatively few cases—with only 265 cases identified across six studies, including the Women’s Health Initiative.93 The estimated RR of endometrial cancer associated with use of cyclic CHT was 1.14 (95% CI = 1.01–1.28),74
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


