Drug resistance and its clinical circumvention
Jeffrey A. Moscow, MD  Kenneth H. Cowan, MD, PhD
Kenneth H. Cowan, MD, PhD  Branimir I. Sikic, MD
Branimir I. Sikic, MD
Overview
Tumors initially sensitive to chemotherapeutic agents frequently develop resistance to them, resulting in the familiar clinical pattern of initial response, followed by a recurrence that no longer responds to therapy. Laboratory and clinical investigations have identified a plethora of drug-resistance mechanisms, some that are particular to an individual agent and others that are generalizable across classes of agents. These mechanisms include altered cellular accumulation and detoxification of drugs, mutation of the drug target, change in expression of the drug target, and activation of alternative signaling pathways. The identification of drug-resistance mechanisms has led to strategies to overcome resistance and improved clinical outcomes.
Systemic therapy with cytotoxic drugs or targeted agents is the basis for most of the effective treatments of disseminated cancers. Additionally, adjuvant chemotherapy can offer a significant survival advantage to selected patients, following the treatment of localized disease with surgery or radiotherapy, presumably by eliminating undetected, minimal, or microscopic residual tumor. However, the responses of tumors to chemotherapeutic regimens vary, and failures are frequent owing to the emergence of drug resistance.
The phenomenon of clinical drug resistance has prompted studies to clarify mechanisms of drug action and to identify mechanisms of antineoplastic resistance. It is expected that through such information, drug resistance may be circumvented by rational design of new noncross-resistant agents, by novel delivery or combinations of known drugs, and by the development of other treatments that might augment the activity of or reverse resistance to known antineoplastics. While earlier mechanisms of drug resistance were identified experimentally by generation of resistant cell lines, recent advances in genomic technology have allowed the direct determination of resistance mechanisms present in clinically resistant tumors.
General mechanisms of resistance to single agents
Experimental selection of drug resistance by repeated exposure to single antineoplastic agents will generally result in cross-resistance to some related agents of the same drug class. This phenomenon is explained based on shared drug transport carriers, drug metabolizing pathways, and intracellular cytotoxic targets of these structurally and biochemically similar compounds.
Generally, the resistant cells retain sensitivity to drugs of different classes with alternative mechanisms of cytotoxic action. Thus, cells selected for resistance to alkylating agents or antifolates will usually remain sensitive to unrelated drugs, such as anthracyclines. Exceptions include emergence of cross-resistance to multiple, apparently structurally and functionally unrelated drugs, to which the patient or cancer cells were never exposed during the initial drug treatment. Despite apparent differences in their presumed sites of action within cells, the drugs associated with multidrug-resistance (MDR) phenotypes frequently share common metabolic pathways or efflux transport systems.
In this section, the processes related to drug resistance will be described. A more comprehensive discussion of selected mechanisms of resistance to specific classes of drugs will be discussed in subsequent sections.
Decreased drug accumulation
Decreased intracellular levels of cytotoxic agents is one of the most common mechanisms of drug resistance. As polar, water-soluble drugs cannot penetrate the lipid bilayer of the cell membrane and require specific mechanisms of cell entry, resistance to these drugs is readily mediated by downregulation of drug uptake mechanisms in tumor cells. For example, antifolates such as methotrexate require specific transporters to gain intracellular access, including high-affinity folate receptors, the reduced folate carrier (SLC19A1), and the proton-coupled folate transporter (SLC46A1) and downregulation of these mechanisms of uptake has been described as causes of methotrexate resistance.1 For hydrophobic, nonpolar drugs that can easily diffuse across the cell membrane, decreased intracellular drug concentrations can be achieved by increasing the activities of drug efflux pumps. For example, overexpression of the P-glycoprotein (MDR1/ABCB1) drug efflux pump is an important example of this mechanism of resistance.2
Altered drug metabolism
Decreased drug activation, increased drug inactivation, or alterations in necessary cofactors can also confer resistance to selected antineoplastic agents. For example, decreased conversion of nucleobase analogs to their cytotoxic nucleoside and nucleotide derivatives by alterations in specific kinases and phosphoribosyl transferase salvage enzymes can lead to resistance to these anticancer drugs.3 Another example associated with resistance is decreased levels of carboxyesterase—an activity necessary to convert a topoisomerase I inhibitor, CPT-11, to its active metabolite, SN-38.4
On the other hand, enhanced inactivation of pyrimidine and purine analogs by increased expression of deaminases is linked to resistance toward these agents.5 Finally, alterations in cofactor levels can also modify drug toxicity. For example, optimal formation of inhibitory complexes between 5-fluorodeoxyuridine monophosphate (FdUMP) and its target enzyme, thymidylate synthase, require the cofactor 5,10-methylene tetrahydrofolate.6 Alterations in the intracellular levels of this cofactor can lead to resistance to fluoropyrimidines.
Increased repair or cellular tolerance to drug-induced damage
Cells contain multiple complex systems involved in the repair of damage to membranes and deoxyribonucleic acid (DNA), and changes in these repair processes can influence drug sensitivity. For example, resistance to cisplatin, a drug whose cytotoxic action involves intrastrand DNA crosslinkages, is associated with increased DNA repair. Conversely, defects in mismatch repair (MMR) are associated with tolerance to cisplatin-induced DNA damage. In this form of platinum resistance, the repair system is apparently unable to recognize platinum-DNA adducts and fails to activate the normal, appropriate programmed cell death response.
Increased rate of detoxification
The manner in which cells metabolize cancer drugs and other xenobiotics is often described as involving three phases of detoxification. Although none of these phases are obligatory steps in the metabolism of every drug, the concept represents a useful framework with which to view cellular detoxification mechanisms. Alterations in any of these three phases can influence the sensitivity or resistance to a particular drug or xenobiotic toxin. Phase 1 drug metabolism is mediated by cytochrome P-450 mixed-function oxidases. Generally, the drug or xenobiotic is rendered into a more electrophilic, reactive intermediate—a process that may enhance toxicity. These metabolites may then be converted to a less-reactive, presumably less-toxic form in phase 2 reactions, which include the formation of drug/xenobiotic conjugations with glutathione (GSH), glucuronic acid, or sulfate—reactions that are catalyzed by multiple isozymes, each of glutathione S-transferases (GSTs), uridine diphosphate (UDP)-glucuronosyl transferases, and sulfatases, respectively. Phase 3 detoxification consists of export of the parent drug/xenobiotic or its metabolites by energy-dependent transmembrane efflux pumps, including MRP (ABCC) family members as described above.
Altered drug targets
Qualitative changes in the enzyme targets of antineoplastic drugs can compromise drug efficacy and have been associated with resistance to targeted agents, especially tyrosine kinase inhibitors (TKIs). These mutations that are acquired through selective pressure often occur in locations that alter the binding site of the TKI. Similarly, other direct targets of enzyme inhibitors develop mutations that overcome resistance to cytotoxic chemotherapy agents, including dihydrofolate reductase (DHFR) resistance to methotrexate, thymidylate synthase resistance to fluoropyrmidines, and topoisomerases I and II resistance to camptothecins.
Altered gene expression
Increased or decreased expression of target enzymes can also lead to drug resistance. These alterations may result from changes that occur at any point along the pathways of gene expression and regulation, including DNA deletion or amplification, altered transcriptional or post-transcriptional control of ribonucleic acid (RNA) levels, and altered post-translational modifications of proteins. In addition, the same molecular mechanisms that lead to oncogenesis can also lead to drug resistance through altered expression of drug targets. Increased expression of DHFR is a mechanism of resistance to methotrexate, while decreased expression of topoisomerase I is a mechanism of resistance to camptothecins.
Activation of alternative signaling pathways
Resistance to targeted agents such as TKIs can be mediated by activation of alternative signaling pathways that provide continued growth stimulation signaling despite successful inhibition of a primary oncogenic event. Activations of several bypass track-signaling pathways have been found in lung cancer patients treated with inhibitors of mutant EGFR and ALK.7 These alternative pathways include amplification of MET, increased HGF expression, PIK3CA mutation, BRAF mutation, and HER2 amplification. The alternative resistance pathways can be identified, and patients treated with relevant inhibitors. In melanomas treated with inhibitors of mutant BRAFV600, mechanisms of resistance include BRAF amplification, increased activity of A-RAF and C-RAF, mutations of NRAS and MEK1, and loss of NF1.8 One strategy for overcoming drug resistance to BRAF inhibitors is concurrent inhibition of more than one kinase in the RAF–MEK–ERK pathway. This strategy was successfully tested in a randomized clinical trial in BRAF-mutated metastatic melanomas of the BRAF inhibitor vemurafenib alone versus vemurafenib plus the MEK inhibitor cobimetinib.9 The combination resulted in a median progression free survival of 9.9 months and remission rate of 68%, compared to 6.2 months and 45% for vemurafenib alone.
Key Points
Mechanisms of resistance to single agents
- Decreased drug accumulation
- Altered drug metabolism
- Increased DNA repair
- Increased detoxification
- Mutation of drug target
- Altered drug target expression
General mechanisms of resistance to multiple agents
Transport-mediated multiple drug resistance (MDR)
De novo and acquired cross-resistance to multiple antineoplastic agents can result from increased expression of a host of promiscuous drug efflux pumps known as ATP-binding cassette (ABC) proteins (Table 1). ABC proteins constitute a large family of 48 transport proteins organized into seven subfamilies, ABCA–ABCG. Of these, at least three have been directly shown to cause MDR, namely MDR1/P-glycoprotein (ABCB1), multidrug resistance-associated protein 1 (MRP1), (ABCC1), and BCRP/MXR/ABC-P (ABCG2). Classic MDR associated with resistance to drugs listed in Table 2 is mediated by P-glycoprotein (MDR1 or ABCB1). A similar but distinct MDR phenotype was attributed to the energy-dependent drug efflux activities of multidrug-resistance protein (MRP) family members, including MRP1 or ABCC1.10
Table 1 ABC transporters associated with multidrug resistance (MDR)
| • P-Glycoprotein/MDR 1/ABCB1-mediated classic MDR |
| • MRP family member-mediated MDR (currently at least three members, MRP1, 2, and 3 in ABCC1, ABCC2, and ABCC3 implicated in MDR drug efflux and detoxification) |
| • BCRP (ABCG2)-mediated MDR (putative ABC half-transporter implicated in mitoxantrone and anthracycline resistance) |
Table 2 Cross-resistance pattern of classic (P-glycoprotein-mediated) MDR
| Class | Drug |
| Anthracyclines | Doxorubicin |
| Daunorubicin | |
| Mitoxantrone | |
| Antibiotics | Actinomycin D |
| Plicamycin | |
| Antimicrotubule drugs | Vincristine |
| Vinblastine | |
| Colchicine | |
| Epipodophyllotoxins | Etoposide |
| Tenoposide |
The genetic basis of acquired MDR has been studied by whole genome sequencing of tumor samples from high grade, serous ovarian cancers. The sequencing disclosed acquired mutations associated with increased expression of the ABCB1 multidrug transporter gene in approximately 8% of patients with recurrent disease after chemotherapy.11 These patients had all been treated with taxanes and/or doxorubicin, which are substrates for the P-glycoprotein transporter. The mutations included fusions at the ABCB1 promoter and translocations in the 5′ region of the gene.
Inhibition of P-glycoprotein during exposure of cancer cells to taxanes such as cabazitaxel has revealed other, alternative mechanisms of resistance to taxanes.12 These alternate mechanisms of resistance include induction of an epithelial to mesenchymal transition (EMT) and increased dynamic instability of microtubules by overexpression of the TUBB3 isoform of beta tubulin.
Another overlapping but discrete MDR phenotype is associated with increased expression of the recently isolated putative efflux transporter, breast cancer resistance protein (BCRP or ABCG2).13 MDR has also been described in association with overexpression of the lung resistance protein (LRP). The mechanism of LRP-associated resistance is unclear, and whether LRP alone is sufficient to confer resistance is unknown. It is speculated that as a major vault protein, LRP is involved in nucleocytoplasmic transport and may be able to prevent entry of drugs into the nucleus.14
Multidrug resistance related to suppression of cell death pathways
Although chemotherapeutic drugs initiate cytotoxicity through their interactions with a variety of molecular targets, cancer drugs affect cell death, at least partially, via downstream events that converge upon pathways mediating type 1 (apoptotic) or type 2 (autophagic) programmed cell death or apoptosis. Apoptosis refers to an orderly cellular death program with predictable molecular and morphologic changes, including nuclear pyknosis and fragmentation, internucleosomal endonucleolytic DNA fragmentation, formation of cytoplasmic apoptotic bodies, and plasma membrane changes, such as transposition of phosphatidylserine to the extracellular surface.15, 16 Autophagy is a pathway for bulk degradation of subcellular constituents that occurs in response to stresses such as nutrient deprivation.17 It involves the creation of autophagosomes/autolysozymes, and can be inhibited by PI3 kinase inhibitors such as 3-methyladenosine and wortmannin.
Although apoptosis may be either TP53-dependent or -independent, frequently the cellular response to DNA damage is regulated by TP53.18 As shown in a simplified diagram (Figure 1), cancer therapy-induced DNA damage is sensed by TP53 by incompletely understood mechanisms. Depending on the particular cell type and damage, TP53 expression may then initiate one of two possible pathways: apoptosis or a process of cell-cycle arrest and repair. In cells where the apoptotic pathway dominates, changes that cause dysfunction or deletion of TP53 are likely to result in reduced apoptosis in response to DNA damage, leading to relative resistance and cell survival with damage.
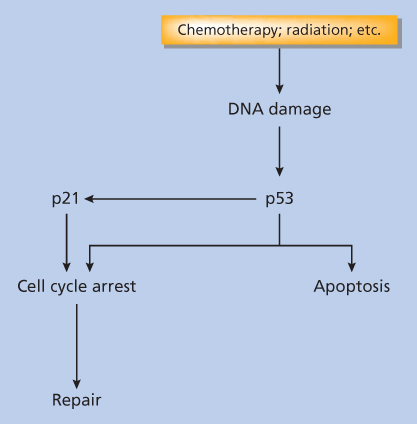
Figure 1 Alternative cellular responses to cancer therapeutic stress.
Other components of cell death-signaling pathways include: the mitogen-activated protein kinase (MAPK)-signaling cascades, which are involved in the regulation of cellular response to exogenous factors, including geno- and cytotoxic cancer treatments; the extracellular stimulus-regulated kinase (ERK1/2) pathway, which is implicated in the proliferative response to growth factors; and the p38 and stress-activated/c-Jun N-terminal protein kinase (SAPK/JNK) pathway. Modulation of these interacting pathways can have a profound effect on whether a cancer cell responds to cytotoxic challenge by activation of apoptosis or by cell-cycle arrest and repair, leading to resistance to treatment.19
Although their mechanism(s) of action is incompletely known, the balance of expressed antiapoptotic family members (Bcl-2 and Bcl-XL) and proapoptotic family members (Bax, Bak, Bad, and Bid) can influence the relative sensitivity of cells to toxic stressors.20 Indeed, increased Bcl-2 and its antiapoptotic homologs are associated with increased resistance of lymphoid cells to the cytotoxic effects of corticosteroids, radiation, and DNA damage from chemotherapeutic drugs.21, 22 It has been proposed that increased levels of antiapoptotic proteins Bcl-2 or Bcl-XL may result in reduced sensitivity to DNA-damaging cancer drugs—a resistance phenotype characterized by cell survival and increased tolerance to DNA damage and genomic instability. This genomic instability may lead to further mutations activating additional resistance mechanisms and conferring more aggressive tumor behavior.20 Although the role of Bcl-2 family proteins and the multidomain proapoptotic members of this family in the development of type 1 (apoptotic) programmed cell death has been extensively characterized, recent studies indicate that these proteins also control type 2 (autophagy) and nonapoptotic programmed cell death.23
The members of the BCL2 family also play important roles in resistance to targeted therapies.24 BIM and PUMA are both proapoptotic BCL2 family members whose expression is induced by targeted kinase inhibitors of oncogenes such as mutated EGFR and BRAF, the BCR-ABL fusion protein, and amplified HER2. Loss of BIM expression has been shown to inhibit apoptosis and results in resistance to inhibitors of these oncogenes. Thus, the expression of mutant and wild-type TP53, Bcl-2 family members, MAPK family members, and other proteins associated with the control of apoptosis and/or autophagy may contribute significantly to the clinical sensitivity of tumor cells. Each of these proteins are the targets of investigational agents.20, 25
Resistance factors due to host-tumor interactions
The failure of chemotherapy to eradicate a tumor in vivo despite exquisite sensitivity to drug in vitro might be a result of anatomic or pharmacologic sanctuaries. For example, the failure to deliver adequate amounts of many drugs across blood–brain and blood–testicular barriers probably accounts for the relatively high frequency of acute lymphoblastic leukemia relapse at these sites prior to addition of CNS-directed therapy to the treatment plans.26 In large solid tumors, chemotherapeutic failures are frequently attributed to decreased drug delivery to a tumor that has overgrown its vascular supply. Additionally, development of acidosis and hypoxia in poorly perfused areas of large tumors may interfere with the cytotoxicity of some drugs. Altered prodrug activation by liver or other normal tissues may profoundly influence the efficacy of drugs such as cyclophosphamide.
Studies by Teicher and Herman27 suggest that tumor–host interactions may influence drug pharmacokinetics and tumor resistance in unexpected ways. In this study, tumor cells selected for cyclophosphamide and cisplatin resistance in vivo were normally sensitive to drugs in vitro. When the tumor cells were reimplanted into nude mice, in vivo drug resistance was restored. These results suggest that resistant tumors may harbor cellular resistance factors that are operative only in conjunction with host factors and, therefore, mediate resistance by altered drug pharmacokinetics in vivo only. If this novel host-dependent mechanism of tumor resistance proves common, these results would provide one explanation for the failure of conventional in vitro testing to predict clinical responsiveness in all cases.
Cancer stem cells and drug resistance
The concept of cancer stem cells (CSCs) developed from observations that individual malignant cells within a cancer differ in their capacity to form tumors. These CSCs, or “tumor initiating cells” form a few to less than 1% of the population, have the property of plasticity or ability to change between tumorigenic and nontumorigenic states, and are a significant factor in the drug resistance of cancers.28 Relapsed cancers are enriched in CSCs, and this subpopulation is known to express high levels of multidrug transporters as well as other drug-resistance genes. At the same time, CSCs offer new targets for cancer therapy, including inhibition of the Notch, Wnt, and Hedgehog pathways.
Genetic basis of acquired drug resistance and tumor heterogeneity
High resolution, deep sequencing of individual cancer genomes has revealed intratumoral heterogeneity of many cancers.29 These data support a branched evolutionary model of tumor growth, with many competing clones differing in growth rates, metastatic potential, and sensitivity to various drugs. There are many implications for anticancer drug resistance that arise from this model. The dominant clone of a cancer derived from a drug-resistant normal tissue, such as colon or kidney, usually reflects the intrinsic resistance of the normal cells from which the cancer is derived. For cancers where the dominant clone is sensitive to a particular drug, the chances are high that drug-resistant minor clones are present at the time of cancer diagnosis. The oncogenic mutations that cause a cancer can contribute to drug sensitivity, by driving cell replication and increasing cancers’ vulnerability to chemotherapy drugs that kill proliferating cells. However, acquired resistance to an anticancer therapy after remission results from resistant clones, some of which may have been present before treatment and some that may have developed during therapy. The emergence of drug resistance is reflected in the disappearance of drug-sensitive clones and overgrowth of resistant clones, an example of Darwinian “survival of the fittest” within cancer populations.30 Heterogeneity in drug-resistance mechanisms and the nature of these mechanisms is also increasingly evident from comparisons of DNA sequences of sequential specimens from individual patients, as well as the evolution of resistance among different patients.31
Deep sequencing is also disclosing differences in genomic instability and apparent rates of mutation among cancers. The factors underlying these differences in genomic instability are not well understood, except for the subset of cancers that have MMR deficiencies. MMR deficiency is associated with a high rate of point mutations, and this may actually lead to more favorable outcomes for immunotherapies that enhance antitumor immunity via immune checkpoint controls (e.g., CTLA4, PD-1, and PDL-1).30 In addition to mutational load, sequencing of melanoma specimens has identified a neo-antigen landscape that was strongly associated with a good response to CTLA-4 blockade by ipilumumab or tremelimumab.32
Both whole genome and targeted genomic sequencing can disclose “actionable mutations” such as BRAFV600E or HER2 amplification that can guide selection of specific targeted drugs, a concept that is being tested in several clinical trials, such as NCI-MATCH (NCT02465060), TRACERx (NCT01888601), DARWIN (NCT02183883), and BATTLE-2 (NCT01248247). Moreover, subclones may be detected that predict eventual failure of a particular therapy, and inform the selection of a noncross-resistant drug active against those subclones.30 The so-called “liquid biopsies” analyzing the circulating free tumor DNA in blood can also detect targetable mutations and the presence of drug-resistant mutations in cancer patients.
In summary, the concept that tumor masses and their metastases were composed of genetically identical clones has given way to a model that pictures each tumor as a tree-like structure with tumor stem cells at the center and multiple evolving clones branching out from the trunk. Therefore, a tumor becomes a heterogeneous collection of related but not identical cells, both in the primary mass and in distant metastases. Selection pressures such as prior therapy can shape the characteristics of the overall tumor cell population in a patient, and spatially separated clones can develop different mechanisms of resistance. This picture of a tumor with its own organism-like qualities has emerged from studies that have performed deep sequencing on multiple biopsies from primary tumor masses and their metastases. The heterogeneity of a tumor extends to both genetic and epigenetic diversity. Thus, strategies to overcome drug resistance must take into account the diversity of mechanisms of resistance to any agent because distinct populations of cells within a patient may harbor different resistance mechanisms.
Key Points
Mechanisms of acquired resistance to multiple agents
- Increased activity of drug efflux pumps
- Suppression of cell death pathways
- Host–tumor interactions
- Quiescent cancer stem cells
- Tumor heterogeneity
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


