Drug Resistance
19.1 INTRODUCTION
A major problem with systemic treatment of cancers is the presence or induction of drug resistance in the tumor cells. In practice many types of cancer that occur commonly in humans (eg, colon cancer, most types of non–small cell lung cancer, pancreatic cancer) have a limited response to treatment with current anticancer drugs. Other human tumors (eg, breast cancer, ovarian cancer, or small cell lung cancer) often respond to initial treatment, but acquired resistance to further therapy usually prevents drug treatment from being curative. Resistance to chemotherapy may have multiple causes, and the most widely studied of these are genetically determined mechanisms that lead to resistance of the individual tumor cells. Sensitivity to drugs may differ widely among cell populations from tumors and normal tissues and also among the cells of a single tumor. The selection or induction of a drug resistant subpopulation in human tumors is a major factor limiting the efficacy of clinical chemotherapy. Even if drug-resistant cells are present initially only at low frequency (eg, 1 drug resistant cell per 105 drug-sensitive cells), their selective advantage during drug treatment will lead to their rapid emergence as the dominant cell population, giving the clinical impression of “acquired resistance.”
There is substantial evidence, reviewed below, that drug resistance may occur through mutation, deletion, or amplification of genes that influence the uptake, metabolism, and efflux of anticancer drugs from target cells. Factors other than genetically determined mechanisms of resistance can lead to clinical resistance of human tumors to anticancer drugs (Sharma et al, 2010). Transient changes in cellular phenotype may occur through epigenetic mechanisms (see Chap. 2, Sec. 2.3): These mechanisms influence the expression of genes (and hence of the proteins encoded by them) as compared to genetic resistance which relates to information transmitted by the DNA sequence of a gene.
The activity of many drugs is dependent on the proliferative status of the cells, and for many of them, on the phase of the cell cycle (see Chap. 17, Sec. 17.5.2). Thus a tumor may appear resistant if many of its constituent cells are nonproliferating or are spared in a drug-resistant phase of the cell cycle. Rapid proliferation of surviving tumor cells (ie, repopulation) between courses of chemotherapy can counter the effects of cell killing and lead to effective resistance. Cure or long-term remission of tumors may be governed by a small population of cells with high proliferative potential (although not necessarily high proliferative rate), so called tumor stem cells (see Chap. 13, Sec. 13.4), and it is the sensitivity of these cells that may ultimately determine success of chemotherapy.
Sensitivity to drugs may depend not only on the intrinsic sensitivity of the constituent tumor cells, but also on the microenvironment and on contact between the tumor cells. Drugs can only exert their lethal effects if they reach the cells at a sufficient concentration to cause lethality. Thus limited vascular access and the requirement to diffuse through tissue from tumor blood vessels are additional causes of drug resistance in solid tumors.
This chapter provides a review of the various mechanisms that lead to clinical resistance of human tumors, as well as potential strategies that might be used to overcome them. These studies are not only important in providing leads for improving therapeutic outcome, but have also contributed to knowledge about the biology of tumors. They have provided information about tumor progression, heterogeneity of the properties of constituent cells, mechanisms of gene regulation and amplification, and mechanisms of transmembrane transport of cellular nutrients and signaling molecules.
19.2 CAUSES OF CELLULAR DRUG RESISTANCE
19.2.1 Molecular Mechanisms of Drug Resistance
A wide range of changes in the properties of tumor cells may lead to resistance to specific drugs; Table 19–1 summarizes some of the underlying mechanisms, which are described in detail in the following sections. Multiple mechanisms of resistance may emerge in response to exposure to a single class of drugs. For example, resistance to several antimetabolite drugs can result from impaired drug uptake into cells, overproduction or reduced affinity of the drug target, upregulation of alternative metabolic pathways, impaired activation or increased inactivation of the antimetabolite, as well as increased drug (or metabolite) efflux. The folic acid analog methotrexate is an example of a drug that can be rendered ineffective by all of these mechanisms (Fig. 19–1; Chap. 18, Sec. 18.3.1). Thus the action of methotrexate depends on active uptake into cells mediated by a membrane transporter, its conversion to more stable intracellular polyglutamated metabolites, and its binding to its target enzyme, dihydrofolate reductase (DHFR), which leads to inhibition of thymidylate and purine biosynthesis, and induction of apoptosis (Fig. 19–1). Similarly, resistance to cisplatin can occur because of changes in processes (drug uptake, drug efflux, and/or intracellular drug sequestration) that prevent adequate levels of the drug reaching its DNA target, or by enhanced activity of processes that repair the DNA after it has been modified by the drug (Fig. 19–2; Chap. 18, Sec. 18.2.2; Kelland, 2007). Because multiple mechanisms may contribute to resistance to every anticancer drug, it is not surprising that initial or acquired drug resistance is observed after treatment of most cell populations.
TABLE 19–1 Cellular mechanisms associated with resistance to anticancer drugs.*
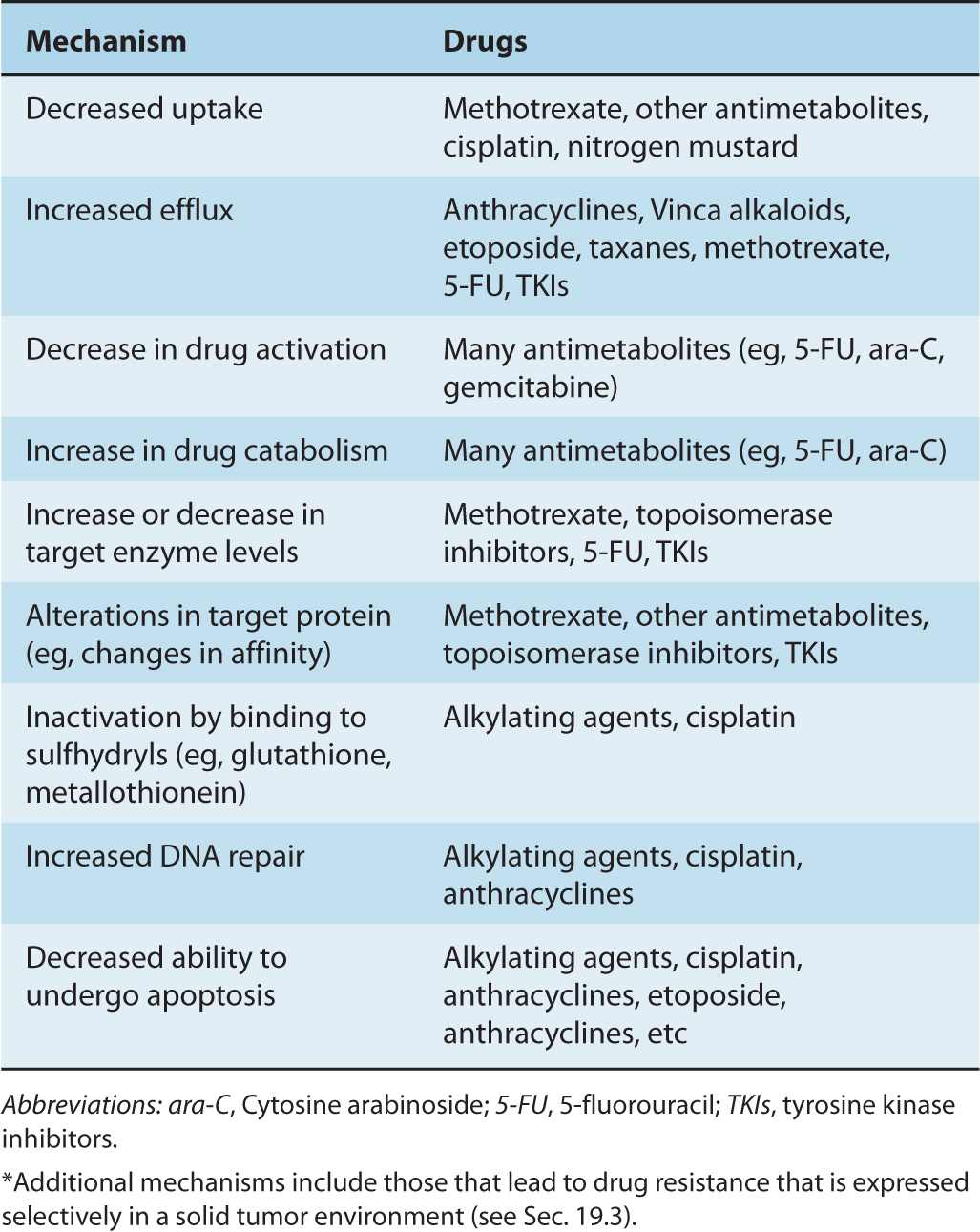
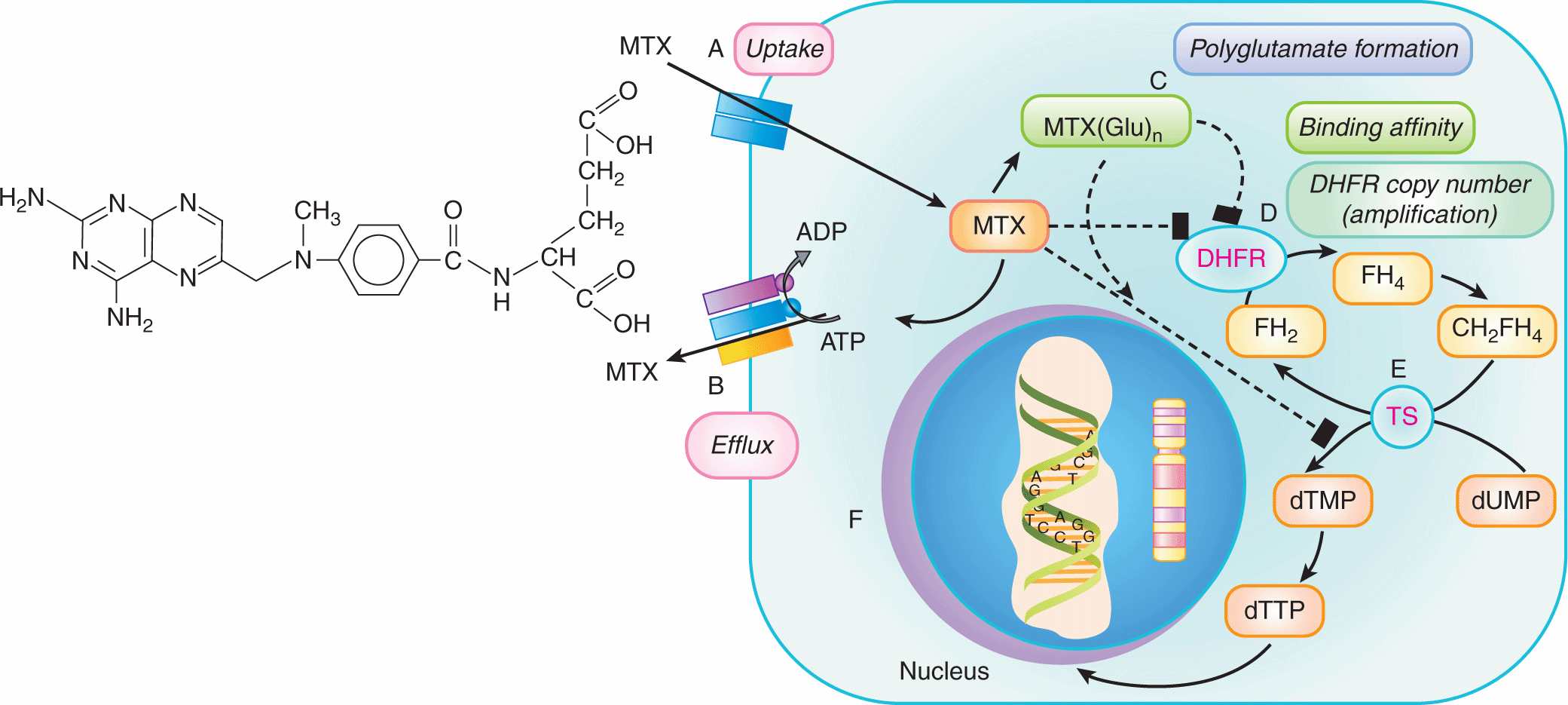
FIGURE 19–1 Multiple molecular mechanisms underlying cellular resistance to methotrexate. Methotrexate (MTX) uptake into cells can be limited by mutations in the reduced folate carrier (RFC1/SLC 19A1) (A). Resistance can also be observed when intracellular levels of MTX are reduced by the action of 1 or more drug efflux pumps such as P-glycoprotein or MRP1 (B). Changes in polyglutamylation of MTX (C) can reduce cellular sensitivity to this drug as MTX and its polyglutamates inhibit the enzyme dihydrofolate reductase (DHFR), causing a block in the conversion of dihydrofolate (FH2) to tetrahydrofolate (FH4), which ultimately results in a reduction in DNA synthesis and cell death. Binding of MTX and its polyglutamates can also be diminished by mutations in DHFR (D). MTX and its polyglutamates inhibit thymidylate synthesis (TS) (E), which also reduces DNA synthesis; drug resistance can occur because of changes in the levels or affinity of this enzyme. Finally, drug resistance may develop when the number of copies of the DHFR gene on chromosome 5 is increased through gene amplification (F).
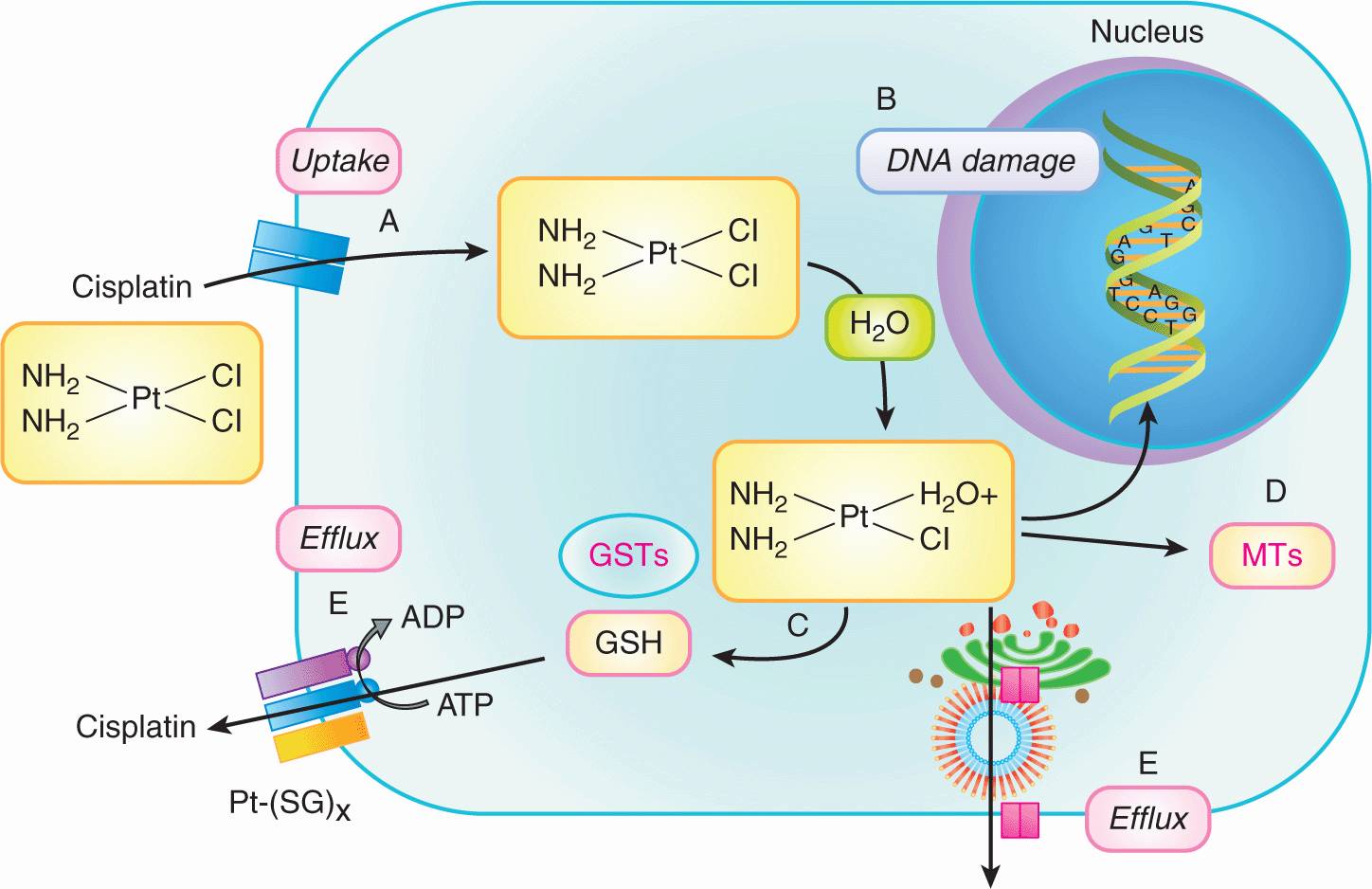
FIGURE 19–2 Multiple molecular mechanisms underlying cellular resistance to cisplatin. Cisplatin uptake into cells can be limited by mutations in the uptake transporter CTR1 (SLC31A1) resulting in drug resistance (A). Once inside the cell, one of the 2 Cl groups is replaced by water producing a reactive nucleophilic species that enters the nucleus where it can covalently modify DNA (primarily by intrastrand binding to adjacent guanines) and cause cell death. Resistance can occur when the damaged DNA is repaired (eg, nucleotide excision repair) or the damaged DNA is “tolerated” (eg, loss of mismatch repair or downregulation of apoptotic pathways) (B). Prior to entering the nucleus, conjugation of the activated cisplatin with glutathione (GSH) by GSH S-transferases (GSTs) (C), or interaction with the sulfhydryl-containing metallothioneins (D) can result in reduced drug efficacy. Finally, resistance can occur if intracellular levels of cisplatin or its metabolites are reduced by the efflux activity of several membrane transporters, including the MRP2 (ABCC2) efflux pump and the ATP7B P-type adenosine triphosphatase (ATPase) transporter (E).
The following evidence indicates that many types of drug resistance are genetic in origin:
1. Characteristics of drug-resistant cells (ie, their pheno-types) are often inherited in the absence of the selecting drug.
2. Drug-resistant cells are generated spontaneously at a rate that is consistent with known rates of genetic mutation.
3. Generation of drug-resistant cells is increased by exposure to compounds that cause mutations in genes or facilitate gene amplification (increased gene copy number). This property has been used to generate and select drug-resistant variant cells that have been used to study resistance phenotypes. Because of the genomic instability of tumor cells and the interaction of many anticancer drugs with DNA, drug treatment may itself accelerate the development of resistance.
4. Altered drug-target proteins that are the products of mutated genes have been identified in many drug-resistant cells.
5. Drug-resistant phenotypes have been transferred to drug-sensitive cells by transfer of genes (see Chap. 2, Sec. 2.2.3).
The presence of drug-resistant cells among the cells in human tumors has implications for planning optimal chemotherapy. Goldie and Coldman (1984) first used mathematical modeling to suggest that the probability of there being at least 1 drug-resistant cell in a tumor population is dependent on tumor size and will increase from near zero to near unity over a small range of tumor sizes (6 doublings), with the critical size depending on the rate of mutation to drug resistance (Fig. 19–3). The Goldie-Coldman model implies a greater chance of cure if therapy is begun early, when only microscopic foci of tumor cells are present, and also predicts a better therapeutic effect when 2 equally effective and “non-crossresistant” drugs are alternated rather than given sequentially, as this minimizes the emergence of cell populations that are resistant to both drugs. Treatment of micrometastatic disease is the principal underlying adjuvant therapy in patients without evident metastatic disease after surgery, which has improved the cure rate for multiple cancers, but it has been difficult to demonstrate benefit from alternate use of non-crossresistant drugs.
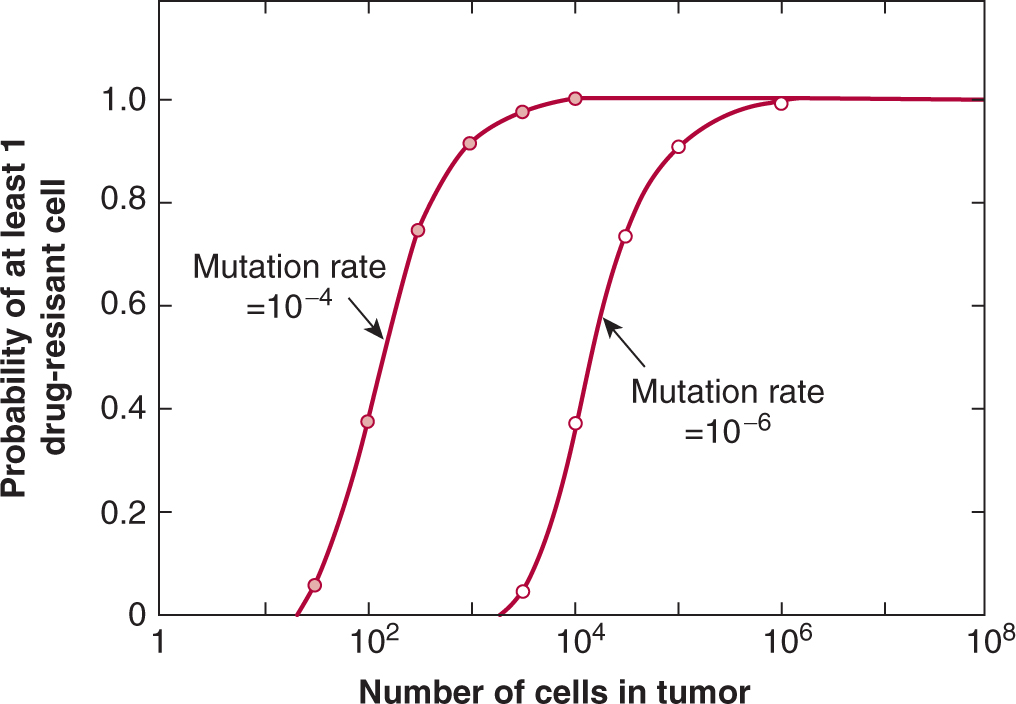
FIGURE 19–3 Probability that there will be at least 1 drug-resistant cell in a tumor containing varying numbers of cells, based on rates of mutation of 10–6 (open symbols) and 10–4 (closed symbols) per cell per generation. Note that this probability increases from low to high values over a relatively short period in the life history of the tumor and that drug-resistant cells are likely to be established prior to clinical detection.
Although drug resistance in many cultured tumor cell lines is caused by gene mutation or amplification, the relevance of these mechanisms to clinical drug treatments is variable. In earlier studies, one method used to select drug-resistant cells was to expose cells to mutagens, followed by selection in high concentrations of drug. This likely predisposed to selection of cells with genetically based drug resistance. However, exposure of cells to lower concentrations of drugs, without prior exposure to mutagens, can also lead to cells that show resistance that may be either stable or transient; transient resistance of some cells in the population may also occur spontaneously, without prior drug exposure.
Mechanisms underlying unstable drug resistance may include transient amplification of genes, changes in patterns of DNA methylation, and other factors that influence gene expression (so-called epigenetic mechanisms). Methylation of cytosines located within CpG dinucleotides is a frequent epi-genetic modification in human DNA (see Chap. 2, Sec. 2.3.2; Sharma et al, 2010). CpG-rich regions (so-called CpG islands) are found typically in the proximal promoter regions of genes, and in normal cells are usually unmethylated. In tumor cells, such regions are more often methylated and transcription of the affected gene may be impaired. Gene inactivation by hypermethylation can have consequences on virtually all pathways in the cell. However, when genes encoding DNA repair enzymes (such as MGMT and hMLH1; Sec. 19.2.6; see Chap. 5, Sec. 5.3), drug transporters (such as the adenosine triphosphate [ATP]-dependent drug efflux transporter ABCG2; Sec. 19.2.3), or proteins that regulate the cell cycle (eg, CDKN2/p16INK4a; see Chap. 9, Sec. 9.2.2) and apoptosis (see Sec. 19.2.8) are hypermethylated, the response to antineo-plastic agents can be markedly altered.
Another epigenetic mechanism of gene regulation that can influence the drug sensitivity of tumor cells is acetylation and deacetylation of histones, nuclear proteins closely associated with DNA (see Chap. 2, Sec. 2.3.1; Muller and Kramer, 2010). Hyperacetylated histones are associated with an open chromatin configuration and they permit transcription of genes to occur. Responses to drugs can be modulated if the gene affected is a known determinant of drug sensitivity. For example, leukemia cells treated with a histone deacetylase (HDAC) inhibitor showed an increased expression of the nuclear drug target, topoisomerase II (see Sec. 19.2.5) and an acquired hypersensitivity to etoposide (Kurz et al, 2001). When combined, inhibitors of DNA methylation and histone acetylation can be particularly effective in restoring drug sensitivity even in solid tumors, at least in experimental systems (Steele et al, 2009). Finally, genes involved in drug sensitivity and resistance can also be regulated by a process known as RNA interference (RNAi). This involves naturally occurring small RNA molecules, known as microRNAs (miRNAs), which can “silence” genes, typically by binding to complementary sequences in the 3′-untranslated regions of target messenger RNA (mRNA) transcripts followed by translational repression or mRNA degradation (Boni et al, 2010; see Chap. 2, Sec. 2.4.3).
19.2.2 Resistance Caused by Impaired Drug Uptake
Drug uptake into cells occurs by one of the following mechanisms: (a) passive diffusion, in which the drug enters the cell by an energy- and temperature-independent process without interacting with specific constituents in the cell membrane; (b) facilitated diffusion, in which the drug interacts in a chemically specific manner with a transport carrier in the cell membrane and is translocated into the cell in an energy- and temperature-independent process; (c) binding of drug to a cell surface receptor that is then internalized; and (d) active transport, in which the drug is actively transported by a carrier-mediated process that is both temperature- and energy-dependent (Sugano et al, 2010). All mechanisms allow for drug entry into cells down a concentration gradient, but the fourth mechanism can also lead to transport against a concentration gradient.
A common mechanism of resistance is impaired unidirectional drug influx. Cellular uptake of hydrophilic drugs is commonly mediated by members of the solute carrier (SLC) superfamily (gene symbol SLC) of membrane transport proteins. This superfamily contains more than 300 proteins organized into 47 families whose major physiological function is to import nutrients and other naturally occurring metabolites into cells.
Many of the SLC importers that have been implicated in the drug sensitivity of malignant and normal cells have a common core structure typified by 2 membrane-spanning domains each containing 6 transmembrane a-helices that form a pore through the membrane. The polytopic SLC proteins mediate the cellular import of a wide range of hydrophilic anticancer drugs that often resemble the natural physiological substrates of the transporter itself. For example, methotrexate is imported across the plasma membrane primarily by an energy-dependent folic acid uptake system, the reduced folate carrier RFC1 (SLC19A1). Drug-resistant cells may have impaired methotrexate uptake into the cell as a result of point mutations in RFC1/SLC19A1 (see Fig. 19–1; Drori et al, 2000); this mechanism of acquired resistance has been found in patients with acute leukemia (Ashton et al, 2009). Similarly, cellular uptake of purine and pyrimidine nucleoside analogs, such as gemcitabine, cladribine, fludarabine, and cytarabine, occurs primarily via membrane transport carriers such as the nucleoside transporters CNT1 and CNT3, which belong to the SLC28 subfamily, and ENT1 and ENT2, which belong to the SLC29 subfamily. Cisplatin and other platinum-containing drugs may be taken up into cells by membrane proteins encoded by the SLC7A11 or CTR1/SLC31A1 genes, which normally import amino acids and copper, respectively. Cells deficient in these carrier proteins are often resistant to these drugs, at least in vitro (see Fig. 19–2; Zhang et al, 2007; Howell et al, 2010). Naturally occurring genetic polymorphisms that result in downregulation or upregulation of these SLC genes in normal and/or tumor cells may also contribute to variation in systemic and intracellular levels of (and hence response to) their drug substrates (Yee et al, 2010).
19.2.3 Multiple Drug Resistance Caused by Enhanced Drug Efflux
Drug accumulation in side cells is determined by the balance between drug uptake and drug efflux. Many anticancer drugs, particularly natural products or their derivatives (eg, doxorubicin, vincristine, etoposide, and paclitaxel), and drug metabolites are effluxed from cells by one or more ATP-binding cassette (ABC) transporters. ABC proteins are found throughout nature from mammals to plants, marine organisms, and prokaryotes, where they carry out many important functions, including the export of potential toxins from cells. Most ABC transporters move one or more molecules (which can range from ions to sugars to small peptides) across biological membranes, a process powered by energy derived from binding and hydrolysis of ATP.
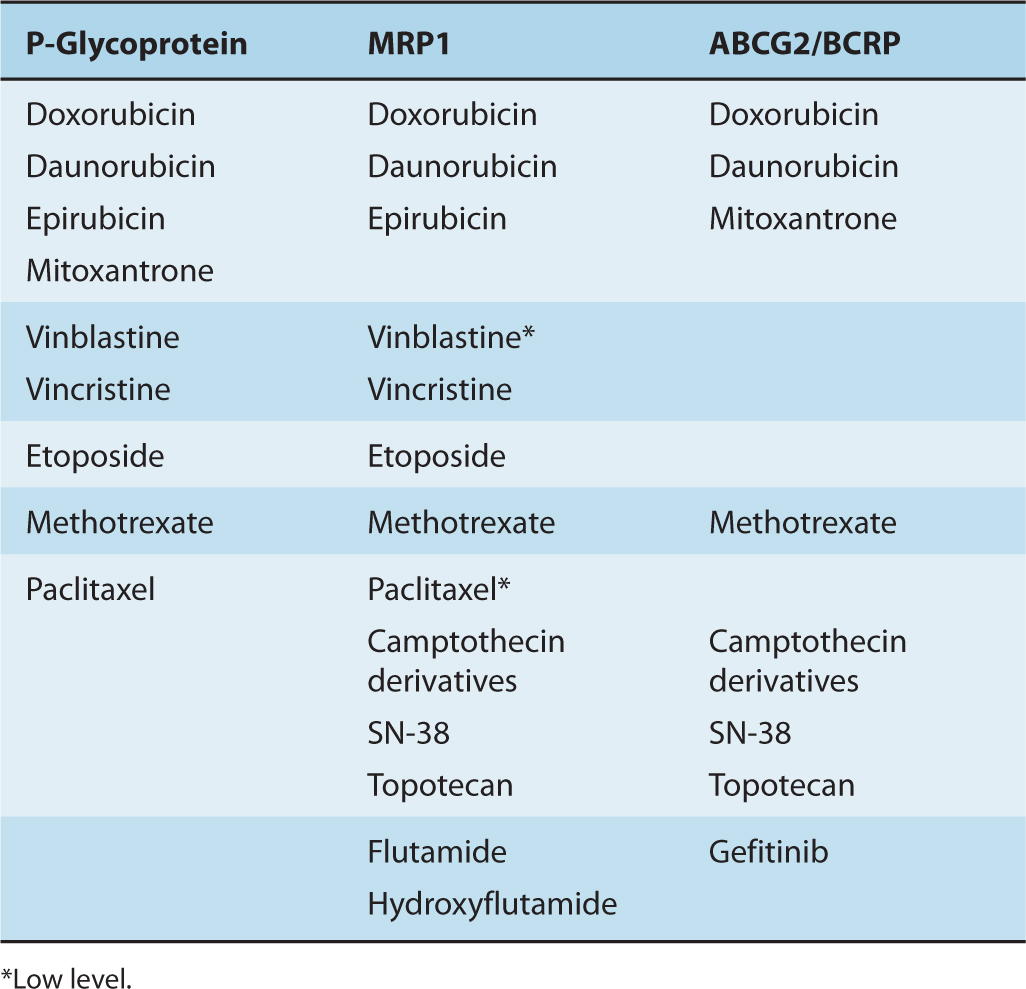
The human ABC superfamily contains 48 proteins, which are organized into 7 subfamilies (A through G) based on the relative similarities of their amino acid sequences (Dean and Allikmets, 2001). Twelve of the 48 ABC transporters are known to efflux drugs and other xenobiotics (or their metabolites) at least in vitro but only 3 of them appear relevant to clinical drug resistance in human tumors and have the ability to export a wide range of structurally diverse anticancer drugs (Table 19–2). The first of these clinically relevant multidrug efflux pumps to be described (in 1976) was P-glycoprotein: human P-glycoprotein is encoded by the ABCB1 gene (formerly known as the MDR1 gene) (Gottesman et al, 2002; Leslie et al, 2005). Multidrug resistance protein 1 (MRP1; gene symbol ABCC1), was described by Cole et al. (1992), while the ABCG2 protein (formerly known as the breast cancer resistance protein [BCRP]) was described by Doyle et al. (1998). These 3 ABC transporters are detected consistently in drug-resistant malignant cells from patients.
TABLE 19–2 ABC transporters that confer multiple drug resistance in human tumors and their specifi city for individual cytotoxic anticancer drugs.
In normal cells, P-glycoprotein, MRP1, ABCG2, and several other clinically relevant MRP-related drug transporters are often expressed in a polarized manner and contribute to drug absorption (bioavailability), distribution (limiting drug access to so-called pharmacological sanctuaries such as the brain, cerebral spinal fluid, and testes), and elimination (efflux into bile or urine) (Fig. 19–4). In contrast to the SLC uptake transporters which generally display specificity for a single class of drugs, the clinically relevant ABC drug efflux pumps typically recognize and transport a structurally diverse array of molecules in addition to the anticancer drugs listed in Table 19–2. It is still not well understood how a single membrane protein can recognize so many structurally dissimilar chemical entities.
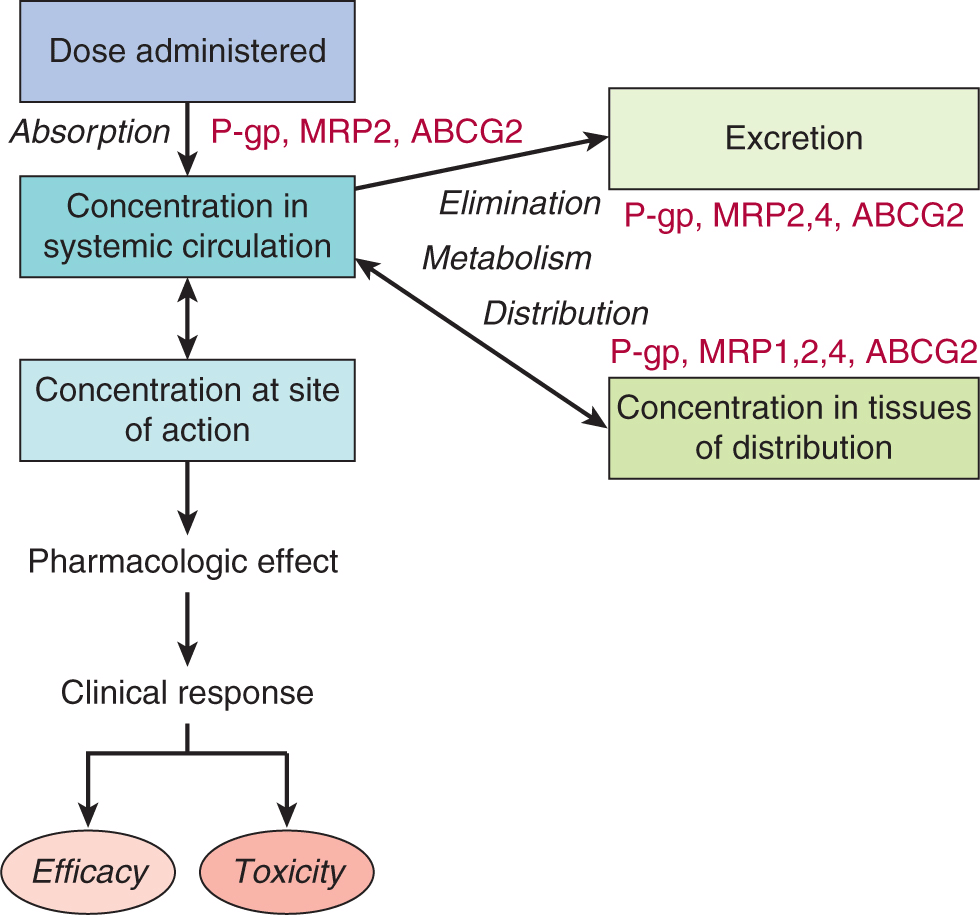
FIGURE 19–4 Multiple roles of the ABC transporters. ABC transport proteins as determinants of drug efficacy and toxicity. While the ABC transporters P-glycoprotein, MRP1, and ABCG2/BCRP have been widely detected in drug-resistant tumor cells, these transporters (together with MRP2 and MRP4) are also now known to affect drug sensitivity and resistance by virtue of their influence on drug absorption (P-glycoprotein, MRP2, ABCG2) and tissue distribution (P-glycoprotein, MRP1, MRP2, MRP4, ABCG2/BCRP), as well as elimination of drugs and their metabolites through the bile, kidney, or other excretory tissue. In tumor cells, the ABC transporters are all found on the plasma membrane. In normal polarized epithelial and endothelial cells in tissues such as the gut, liver, kidney, and brain which are important for drug absorption, distribution and elimination, P-glycoprotein, MRP2, and ABCG2 are found on apical membranes while MRP1 is found on basolateral membranes. MRP4 is unusual in that its localization depends on the tissue in which it is expressed (eg, apical in kidney, brain; basolateral in prostate, liver). P-gp, P-glycoprotein. (Slot et al, 2011).
19.2.3.1 P-Glycoprotein In humans, ABCB1 (formerly MDR1) has been mapped to chromosome 7, and its gene product, P-glycoprotein, is a relatively large phosphoglycoprotein of molecular weight 170 kDa. It contains 2 homologous halves, each of which is comprised of 6 transmembrane α-helices followed by a cytosolic nucleotide (ATP)-binding domain. Thus, like most ABC proteins, P-glycoprotein has a typical 4-domain structure (Fig. 19–5A) and all 4 domains are required for its full activity. The drug-binding sites of P-glycoprotein are generally found in the 2 hydrophobic membrane-spanning domains, which together also form the drug translocation pathway through the membrane (Fig. 19–5A). Structural evidence indicates that for transport to occur, these hydrophobic domains are coupled with the 2 energy-providing ATP-binding domains (NBD1/2), and this coupling is mediated by the cytoplasmic loops that connect the transmembrane helices to one another. The details by which this coupling of substrate transport to the adenosine triphosphatase (ATPase) activity of these drug efflux pumps occurs are not completely understood but are thought to be common among all ABC proteins (Hollenstein et al, 2007).
P-glycoprotein confers resistance against a wide spectrum of complex heterocyclic hydrophobic, antineoplastic drugs mostly derived from natural products that include the anthracycline antibiotics, the Vinca alkaloids, and the taxanes (see Table 19–2). Other substrates include the tyrosine kinase inhibitors gefitinib and imatinib (see Chap. 17, Sec. 17.3.1). P-glycoprotein also mediates the cellular efflux of other drugs used with cancer chemotherapy, such as ondansetron and granisetron, that are used to control emesis. Certain fluorescent chemicals (eg, rhodamine 123) and radiopharmaceuticals used in imaging (eg, 99mTc-sestamibi) are also P-glycoprotein substrates and are being investigated for their ability to detect clinical drug resistance mediated by this transporter, the latter by external imaging.
In vitro studies show that single amino acid substitutions can markedly alter the substrate specificity of P-glycoprotein (Loo and Clarke, 2005). Furthermore, a naturally occurring polymorphism in the ABCB1 gene has been described that affects the ability of P-glycoprotein to recognize some of its drug substrates (Kimchi-Safarty et al, 2007).
In addition to being expressed at elevated levels in certain tumor types, P-glycoprotein is also found in normal tissues, such as the kidney and adrenal gland, as well as the lung, liver, and gastrointestinal tract. P-glycoprotein is localized to the apical surface of polarized cells that line the tubules or ducts or lumen of these organs, and thus provides such cells with a mechanism for extruding xenobiotic molecules that are recognized by the transporter, or for impeding uptake of these molecules (delaying absorption) (see Fig. 19–4; Leslie et al, 2005). P-glycoprotein is also expressed on the apical membrane of endothelial cells lining the blood–brain barrier, where it excludes toxic natural products from the central nervous system. Strong evidence for the function of P-glycoprotein as a regulator of drug uptake comes from studies of mice in which these genes have been disrupted by homologous recombination (see Chap. 2, Sec. 2.4.5). These P-glycoprotein Abcb1 knockout mice display a marked increase in sensitivity to the neurotoxic side effects of several different drugs (Lagas et al, 2009). Such animals also show enhanced oral absorption (bioavailability) of certain drugs, and this appears also to be true in humans when P-glycoprotein is inhibited.
Many investigators have measured levels of P-glycoprotein in human tumors, both before and after treatment with anticancer drugs (Gottesman et al, 2002). Elevated P-glycoprotein has been found in sarcomas and in cancers of the colon, adrenal, kidney, liver, and pancreas. All these tumors tend to be resistant to chemotherapy. Elevated levels of P-glycoprotein have also been detected following relapse after chemotherapy in more drug-sensitive tumors, including multiple myeloma and cancers of the breast and ovary. These findings suggest that P-glycoprotein may contribute to clinical drug resistance. Increased P-glycoprotein has also been reported to correlate with a poor prognosis in children with neuroblastoma, rhab-domyosarcoma, and osteogenic sarcoma.
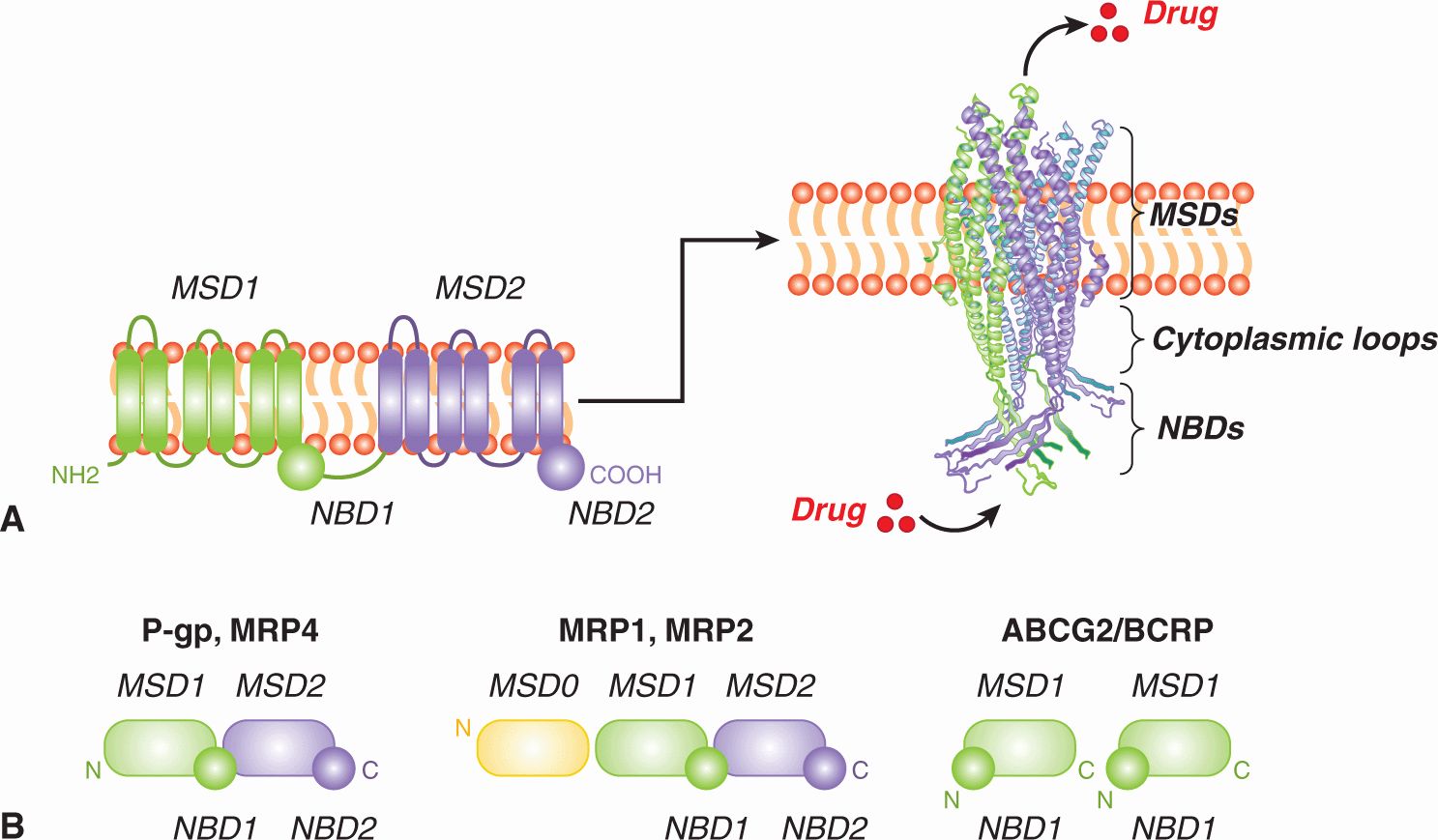
FIGURE 19–5 General structure of ABC membrane drug efflux pumps. A) (left) Shown is a linear topological cartoon of the core structure of ABC transporters such as P-glycoprotein showing the 2 cytoplasmic nucleotide-binding domains (NBDs) and 12 transmembrane (TM) helices (here shown as cylinders) equally distributed between 2 membrane-spanning domains (MSDs). (right) Shown is a 3D homology model of the core structure of MRP1 (lacking MSD0, see Fig 19–5B) generated using the crystal structure of Staphylococcus aureus Sav1866 as template (Hollenstein et al, 2007; DeGorter et al, 2008). The a-carbon backbone in ribbon representation of the core structure (MSD1-NBD1-MSD2-NBD2) is viewed from the plane perpendicular to the membrane bilayer (DeGorter et al, 2008). Homology models of P-glycoprotein, MRP4 and other ABC transporters look very similar. The 2 NBDs form a “sandwich” dimer for the effective binding and hydrolysis of 2 molecules of ATP, providing the energy for the transport process. Signaling between the MSDs (translocation pathway through the membrane) and the NBDs (which provide the energy for transport) is mediated by specific sequences in the cytoplasmic loops. Substrates that enter the cell by diffusion or active transport, or are formed in the cell by conjugation, are thought to be exported from the cell either directly through the pore from the cytoplasm, or in the case of hydrophobic drugs, are taken up from the inner leaflet of the membrane lipid bilayer. B) Domain organization of ABC transporter drug (and drug metabolite) efflux pumps implicated in drug resistance in malignant cells. P-glycoprotein and the MRPs are encoded as multidomain single polypeptides containing MSDs and NBDs in the orientations shown, while ABCG2/BCRP is encoded as a “half-transporter” and 2 identical subunits assemble together to form a functional transporter. Each of the MSDs contains 6 transmembrane segments (a-helices) except for MSD0 of MRP1 and MRP2 which contains just 5. P-gp, P-glycoprotein.
19.2.3.2 Multidrug Resistance Proteins A second multidrug transporter, now known as MRP1 (gene symbol ABCC1), was cloned originally from a drug-selected cell line derived from human small cell lung cancer that did not express P-glycoprotein but did contain multiple gene copies of ABCC1 (Cole et al, 1992; Fig. 19–6). Although P-glycoprotein and MRP1 are both members of the ABC superfamily, the 2 proteins share only 15% amino acid sequence identity and differ in several significant structural and pharmacological ways (Leslie et al, 2005). The ABCC1 gene is located on chromosome 16p13.1 and encodes a 190-kDa phosphoglycoprotein with 17 transmembrane a-helices, 5 more than P-glycoprotein. These extra transmembrane helices of MRP1 form a third NH2-proximal membrane-spanning domain, MSD0 (see Fig. 19–5B). The role of this NH2-terminal hydrophobic extension has not been elucidated, but it appears important for the transport of some MRP1 substrates and has been implicated in stabilizing expression of this transporter at the plasma membrane (Deeley et al, 2006).
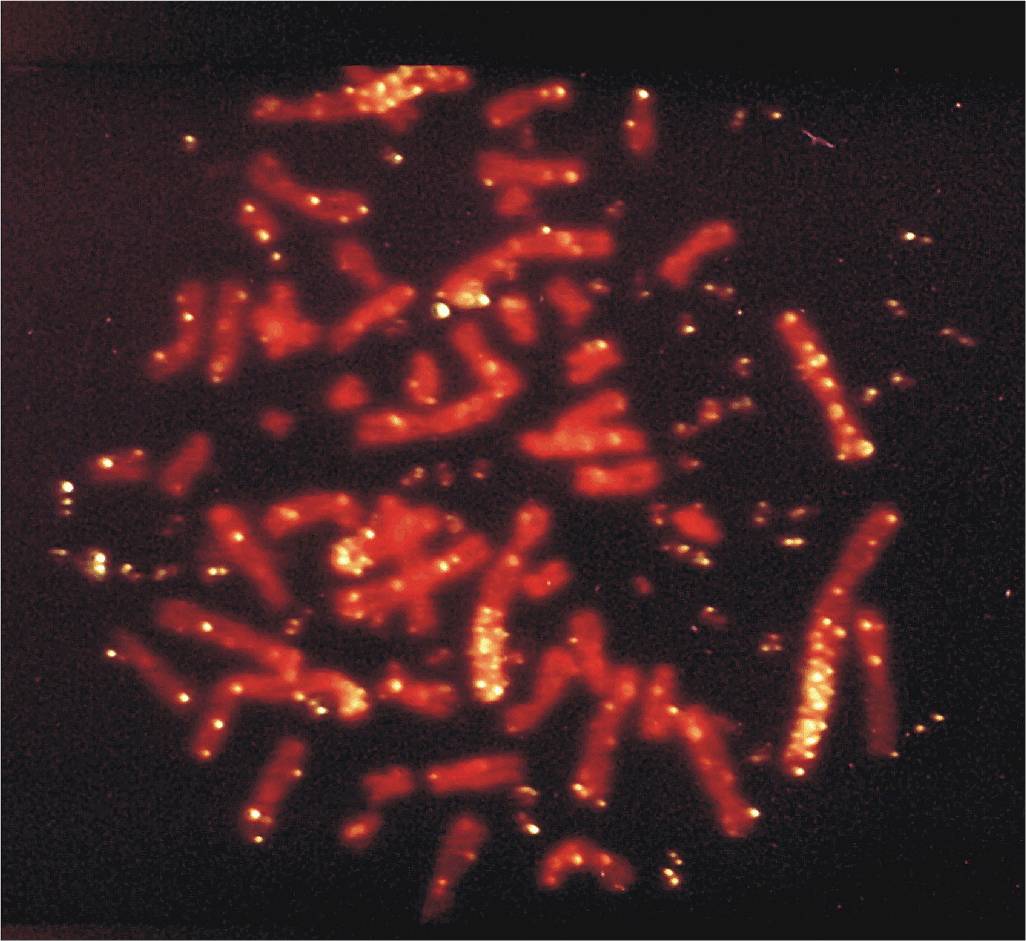
FIGURE 19–6 Metaphase spread of a highly drug-resistant lung cancer cell that contains approximately 100 copies of the ABCC1 gene. Fluorescent in situ hybridization analysis of ABCC1, the gene encoding the drug efflux pump MRP1. The normal cellular locus of ABCC1 is chromosome 16p13.1. However, in many drug-resistant cell lines where expression of MRP1 is elevated, ABCC1 has been amplified. The figure shows a metaphase spread of a highly drugresistant lung cancer cell that contains approximately 100 copies of the ABCC1 gene. Note that the fluorescently labeled ABCC1 probe has hybridized to several homogeneously staining regions (HSRs) and multiple double minute chromosomes (DMs). (From Slovak et al, 1993).
As for P-glycoprotein, increased expression of MRP1 leads to a net decrease in cellular accumulation of a variety of anticancer drugs, including both natural products and the folic acid analog methotrexate (see Table 19–2). The spectrum of drugs that MRP1 transports is slightly different from P-glycoprotein in that MRP1 confers at most low levels of resistance to the hydrophobic paclitaxel and vinblastine. Also, transport of some drugs (eg, vincristine, daunorubicin) by MRP1 depends on the presence of the antioxidant glutathione (GSH) or a tri-peptide analog (Fig. 19–7A; Rappa et al, 1997; Loe et al, 1998). In vitro, GSH causes changes in the conformation of the MRP1 protein, which increases its affinity for some of its drug substrates (Cole and Deeley, 2006; Rothnie et al, 2006), but GSH may also influence MRP1 function in other ways.
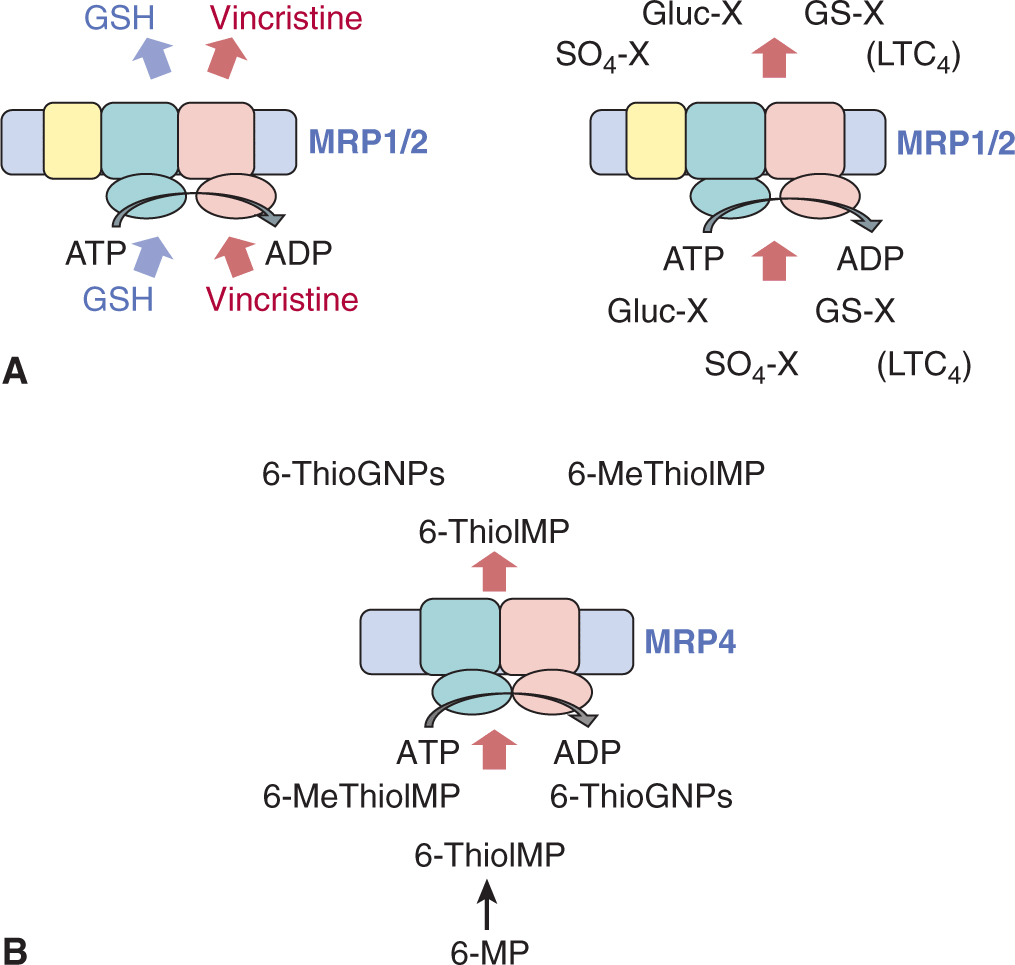
FIGURE 19–7 Transport of conjugated and unconjugated drugs by “long” and “short” MRPs across cell membranes. A) Efflux of several natural product drugs (eg, vincristine and doxorubicin) by the “long” MRP1 or MRP2 is dependent on the presence of reduced GSH, which is cotransported with the drug (left). Transport of drug metabolites, including those conjugated to glutathione (GS-X), glucuronide (Gluc-X), and sulphate (SO4-X) (right) by MRP1 (and MRP2) usually does not require GSH. The major physiological metabolite transported by MRP1 is the GSH-conjugated leukotriene C4 (LTC4). B) Efflux of the active metabolites of 6-mercaptopurine (6-MP) by the “short” MRP4.
In addition to its ability to confer resistance to anticancer drugs in both malignant and normal cells, MRP1 transports a broad spectrum of organic anions, another property not shared by P-glycoprotein. For example, metabolites of drugs and other xenobiotics that are conjugated to GSH, glucuronide, or sulfate (the products of Phase II drug metabolism; see Fig. 19–7A), are frequently effluxed by MRP1 but not by P-glycoprotein. Conjugates of endogenous metabolites are also transported by MRP1, most notably the cysteinyl leukotriene, LTC4, a potent mediator of inflammation. Thus, mice bearing a disrupted Abcc1 gene exhibit enhanced sensitivity of tissues such as seminiferous tubules and the oropharyngeal cavity to anticancer agents like etoposide, but also an impaired response to proinflammatory stimuli associated with diminished LTC4 secretion from mast cells (Wijnholds et al, 1997; Lagas et al, 2009). Other potential physiological substrates of MRP1 include the conjugated estrogens estradiol glucuronide and estrone sulfate, folic acid, GSH and its prooxidant metabolite GSH disulfide (GSSG) (see Fig. 19–7A). MRP1 also transports several fluorescent organic anions, such as calcein, which facilitates measurement of the activity of this transport protein in clinical and experimental studies. Thus the substrates of MRP1 are more diverse than those of P-glycoprotein (Leslie et al, 2005; Slot et al, 2011).
MRP1 has been detected in a wide variety of human tumors and normal tissues (Deeley et al, 2006). Increased expression has been observed in several types of drug-resistant human tumors, such as lung cancer and some leukemias, and in many cell lines derived from human tumors. In children with neuroblastoma, expression of MRP1 was correlated with expression of the N-MYC oncogene and predicted poor survival (Haber et al, 2006). In vitro, certain mutations in ABCC1 cause changes in the substrate specificity of MRP1, such as substitution of Trp1246, which results in total loss of drug resistance (Ito et al, 2001). In primary neuroblastoma, the naturally occurring ABCC1 polymorphism G2012T is associated with patient outcome and altered stability of the ABCC1 gene transcript (Pajic et al, 2011).
Eight additional MRP-related proteins have been described, 5 of which have been shown to transport one or more drugs, at least in vitro, but have no significant role in tumor cell resistance (Deeley et al, 2006; Slot et al, 2011). Despite the fact it is rarely found in tumors, however, MRP2 in normal cells may impact drug sensitivity because it can influence the distribution and elimination (and hence pharmacokinetics) of some anticancer drugs and their metabolites (see Fig. 19–4). MRP2 is structurally very similar to MRP1, but unlike MRP1, it is expressed predominantly on apical membranes of the bile canaliculus, renal epithelium, and intestinal enterocytes. Thus, MRP2 plays a role in the oral bioavailability and elimination of drugs and their metabolites that are substrates of this transport protein (Table 19–3; Nies and Keppler, 2007).
MRP4 (gene symbol ABCC4) is a “short” MRP because it lacks the third NH2-terminal membrane-spanning domain of MRP1 and MRP2 (Slot et al, 2011) and thus is a 4-domain ABC transporter like P-glycoprotein. MRP4 can transport topotecan and the monophosphorylated, bioactive forms of the antimetabolites 6-mercaptopurine and 6-thioguanine, as well as nucleotide analogs used to treat patients with viral infections (Leggas et al, 2004; Russel et al, 2008; see Fig. 19–7B and Table 19–3). However, like MRP2, increased levels of MRP4 have only rarely been detected in tumor samples from patients. In contrast, a role for MRP4 in drug disposition/elimination (pharmacokinetics) and adverse drug reactions has been revealed by studies of Abcc4-/- mice. Topotecan accumulation in both brain tissue and in cerebrospinal fluid is enhanced in Abcc4-/- mice, reflecting the unusual dual localization of MRP4 at the basolateral membrane of the choroid plexus epithelium, and at the apical membrane of the endothe-lial cells of the brain capillaries (Leggas et al, 2004). Renal elimination of many drugs (or their metabolites) is also reduced in Abcc4-/- mice, consistent with a protective role for MRP4 in the kidney. Common nonsteroidal antiinflammatory drugs (NSAIDs) (eg, celecoxib) inhibit transport by MRP4, which may contribute to renal toxicity when a cytotoxic agent such as methotrexate is coadministered with NSAIDs (El-Sheikh et al, 2007). Abcc4-/- mice are also more sensitive to the hematopoietic toxicity of thiopurines. In humans, a polymorphism in ABCC4 encodes a nonfunctional MRP4 transporter and the generally greater sensitivity of Japanese patients to thiopurines may reflect the greater frequency (>18%) of this polymorphism in the Japanese population (Krishnamurthy et al, 2008).
TABLE 19–3 Drugs and drug metabolites that interact with MRP2 and MRP4 that may be important for drug disposition and elimination.

19.2.3.3 ABCG2 The third ABC drug efflux pump of clinical relevance in drug resistance of tumor cells is ABCG2 (formerly BCRP), which was first cloned from a drug-resistant breast cancer cell line that expressed neither P-glycoprotein nor MRP1 (Doyle et al, 1998). The ABCG2 gene has been mapped to chromosome 4q22 and encodes a protein containing just 655 amino acids, compared to 1280 amino acids for P-glycoprotein and 1531 amino acids for MRP1. ABCG2 is comprised of only a single membrane-spanning domain and nucleotide-binding domain, and is often referred to as a “half-transporter”: 2 ABCG2 proteins must come together to form a functional 4-domain transporter (see Fig. 19–5B).
When overexpressed, ABCG2 can render tumor cells resistant to a variety of clinically important drugs, including mitoxantrone, doxorubicin, daunorubicin, topotecan, and SN-38 (the active metabolite of irinotecan), as well as the epidermal growth factor receptor (EGFR) inhibitor gefitinib (see Table 19–2) (Doyle et al, 1998; Vlaming et al, 2009). Thus despite substantial differences in their amino acid sequences and structural organization, ABCG2 can efflux some of the same drugs as P-glycoprotein and MRP1. Like P-glycoprotein and MRP1, ABCG2 also transports some fluorescent molecules, which facilitates detection of this transporter in clinical samples.
ABCG2 is widely expressed in normal tissues and, like P-glycoprotein and MRP2, is present on apical membranes of polarized endothelial and epithelial cells, and has been implicated in the absorption, distribution and elimination of certain xenobiotics. In a mouse model, Abcg2 has been shown to contribute to the blood–brain, blood–testis, and blood–fetal barriers (Vlaming et al, 2009). Efflux of the fluorescent dye Hoechst 33342 has been reported in the so-called side-population of bone marrow cells that are highly enriched for undifferentiated stem cells. This ABC transporter might serve to extrude a metabolite(s) from embryonic stem cells that helps prevent differentiation (Sarkadi et al, 2010). ABCG2 likely also serves a protective role in these cells against damage by xenobiotics.
19.2.3.4 Reversal or Circumvention of Drug Resistance Mediated by ABC Transporters Many agents have been identified that inhibit the function of P-glycoprotein and increase the sensitivity of drug-resistant tumor cells in culture (eg, Gottesman et al, 2002). Some of these agents are themselves substrates for P-glycoprotein and competitively inhibit the efflux of anticancer drugs, but non-competitive mechanisms have also been implicated. Multiple clinical trials have assessed the potential of P-glycoprotein antagonists to increase the sensitivity of human tumors to anticancer drugs such as doxorubicin and vinorelbine. Some patients with hematological malignancies that were drug resistant responded to the same anticancer drugs when an inhibitor of P-glycoprotein was added to the drug regimen, but the results of studies with solid tumors have been disappointing. There are several possible reasons for the inconclusive outcomes of many of these clinical trials (Gottesman et al, 2002; Yu et al, 2012), including poor trial design, resistance because of mechanisms in addition to or other than P-glycoprotein, and failure to achieve adequate levels of the reversing agent in the tumor tissue. Several “third-generation” P-glycoprotein reversal agents with higher affinity and greater potency and specificity are under investigation.
Agents such as verapamil and cyclosporine, which may reverse drug resistance as a result of P-glycoprotein in cell culture, have much less effect on drug resistance because of MRP1. The cysteinyl leukotriene receptor antagonist MK-571 was identified as a relatively potent inhibitor of MRP1-mediated transport, but suffers from a lack of specificity. High-throughput screening has identified more potent and specific agents that antagonize the drug efflux activity of MRP1, but these have not yet been widely tested in humans (Boumendjel et al, 2005; Burkhart et al, 2009). In cultured cells, resistance to some (but not all) drugs caused by MRP1 can be reversed by using agents that deplete cellular GSH, but it is not yet known if this is the case in humans (Cole and Deeley, 2006).
The search for modulators of drug efflux mediated by ABCG2 has been less extensive, although several highly specific inhibitors have been identified by high-throughput screens of large chemical libraries (Vlaming et al, 2009). Although these agents have been shown to be effective in vitro and in mice bearing drug-resistant human tumors, none thus far have been tested in clinical trials.
Novel agents that inhibit the transport function of P-glycoprotein (as well as MRP1, MRP2, MRP4, and ABCG2) are also being investigated for their ability to improve oral absorption of antineoplastic drugs or enable better penetration of drugs into pharmacological sanctuaries (eg, the central nervous system), tissues that are normally protected by these transporters (Leslie et al, 2005; Matsson et al, 2009). These studies reflect a growing appreciation of the role that transporters in normal cells may play in the clinical efficacy and toxicity of drugs as well as drug–drug interactions (see Fig. 19–4). The ability of these ABC proteins to confer drug resistance has also led to exploration of the use of vectors to deliver genes encoding these transporters into bone marrow and other drug-sensitive normal tissues to protect them from the toxic side effects of chemotherapy.
19.2.4 Resistance Caused by Decreased Drug Activation or Increased Drug Inactivation
Many antineoplastic drugs, and in particular the antimetabolites, must be converted to a pharmacologically active form after cellular uptake in order to exert their cytotoxic effects (see Chap. 18, Sec. 18.3). Resistance to these agents can occur when there is a decrease in activity or levels of the activating enzyme(s), or an increase in the activity or levels of an enzyme that is responsible for detoxifying the active form of the drug. For example, drug-resistant leukemia cells may show a decrease in polyglutamylation of intracellular methotrexate as a result of either decreased activity of the synthetic enzyme, folylpolyglutamate synthase, or increased activity of the catabolic enzyme, folylpolyglutamate hydrolase (see Fig. 19–1). Resistance to the pyrimidine analog cytosine arabinoside (ara-C) may occur as a result of decreased activation by various kinases, such as deoxycytidine kinase and/or enhanced inactivation by deaminases. Similarly, resistance to the pyrimidine analog, 5-fluorouracil (5-FU), is associated with alterations in the enzymes (eg, uridine monophosphate [UMP] kinase) responsible for its activation (Humeniuk et al, 2009), or of its catabolism (eg, dihydropyrimidine dehydrogenase; Yang et al, 2011).
Many anticancer drugs and carcinogens cause cellular damage by the production of chemically reactive electro-philic intermediates, especially reactive oxygen species (see Chap. 4, Sec. 4.2.3). Similar processes are involved during the interaction of ionizing radiation with tissue (see Chap. 15, Sec. 15.2.3). One mechanism by which cells can protect themselves from damage caused by reactive agents is by upregulating the synthesis of sulfhydryl-containing molecules, especially the nucleophilic tripeptide GSH, which can form conjugates with the electrophilic metabolites and render them less reactive and thus nontoxic. The importance of GSH in the protection of normal cells is reflected in its widespread distribution and its relatively high intracellular concentration ( >1 mM in many tissues). GSH can inactivate peroxides and free radicals, which may be produced by drugs such as etoposide and the anthracyclines (eg, doxorubicin). It can also react with positively charged electrophilic molecules, such as the active groups of cisplatin and alkylating agents, rendering them less toxic and more easily excreted. These reactions are catalyzed, respectively, by the enzymes GSH peroxidase and GSH S-transferase (GST) (see Fig. 19–2).
By conjugating GSH to various drugs or their active metabolites, GSTs appear to play a role in the development of cellular resistance to some antineoplastic agents (Townsend and Tew, 2003; Sau et al, 2010). The cytosolic GSTs are a highly polymorphic multigene family of enzymes that are often classified by their isoelectric points, as well as by their relative sequence homology: the major classes are the basic (α class), neutral (μ class), and acidic (π class) GSTs. Each functional GST enzyme is a homo- or heterodimer made up of subunits encoded by gene loci from within a given class. Not all cell lines selected for resistance to alkylating agents have shown increases in GST protein levels or activity, but several lines of evidence support a role for GSTs in resistance to alkylating and platinum-containing agents:
1. Nitrogen mustards can form GSH conjugates in reactions catalyzed by GSTs.
2. Human tumors and tumor cell lines often overexpress GST isozymes.
3. GST inhibitors can sometimes sensitize cultured tumor cells to cisplatin and alkylating agents (Pasello et al, 2008; see Fig. 19–2).
4. Cell-cycle-dependent sensitivity to melphalan correlates with the cell-cycle–dependent expression of certain GSTs.
5. Transfection of complementary DNAs (cDNAs) encoding certain GST isoforms can confer resistance to alkylating agents (Smitherman et al, 2004).
6. Elevation of GST can occur within several days of exposure to chlorambucil as part of the normal cellular response.
In addition to their conjugating activities, the n and |L classes of GSTs have an antiapoptotic function because of their regulatory role in the mitogen-activated protein (MAP) kinase pathway via inhibitory interactions with c-Jun N-terminal kinase 1 (JNK1) and ASK1 (apoptosis signal-regulating kinase) (Townsend and Tew, 2003; Sau et al, 2010; see Chap. 8, Sec. 8.2.4). Thus GSTs may contribute to resistance by facilitating drug detoxification as well as by acting as an inhibitor of the MAP kinase pathway.
Drugs conjugated to GSH, glucuronide, or sulfate groups are negatively charged and these conjugated organic anions are extruded from cells by an energy-dependent process. GSH-conjugate export carriers (known variably as GS-X pumps or multispecific organic anion transporters) are involved, and this export function is undertaken, in large part, by the ABC transporters MRP1 and MRP2 (Cole and Deeley, 2006; Sec. 19.2.3). Although conjugated metabolites are usually less reactive, the active efflux of conjugated metabolites by the MRP transporters prevents their intracellular accumulation, thereby reducing the possibility of hydrolytic enzymes causing the regeneration of the active parent compound. However, some conjugated metabolites can be directly cytotoxic because of their ability to inhibit enzymes important for cell viability, as well as by inhibition of the conjugating enzymes. Thus, elimination of conjugated metabolites from the cell is an important component of the detoxification process (Leslie et al, 2005).
Resistance to cisplatin and alkylating agents also is associated with increased levels of metallothioneins, proteins rich in sulfhydryl-containing cysteine residues (see Fig. 19–2). The presumed mechanism is “neutralization” of the toxic electro-philic drugs or their metabolites, by their interaction with these proteins; indeed, each molecule of metallothionein can bind up to 10 platinum atoms. Nevertheless, although cells transfected with a human metallothionein gene can acquire resistance to cisplatin and alkylating agents (Kelley et al, 1988), and cells that do not produce metallothionein show increased chemosensitivity (Kondo et al, 1995), there is no in vivo evidence to support a major role for metallothioneins in clinical drug resistance.
19.2.5 Resistance Caused by Altered Levels or Modification of the Drug Target
To exert their cytotoxicity, antineoplastic agents must interact efficiently with their intracellular protein targets. Changes may occur such that levels of the target protein are increased, thus requiring increased concentrations of drug to elicit cytotoxicity. Alternatively, the gene encoding the protein can acquire a mutation such that it retains its normal physiological activity but exhibits reduced affinity for drugs. These resistance mechanisms reduce the effectiveness of both conventional (below) and novel targeted therapeutic agents (see Sec. 19.2.7).
19.2.5.1 Resistance to Drugs Targeting Enzymes Involved in Folate Metabolism and DNA Synthesis Among the multiple mechanisms of resistance to 5-FU, is the acquisition of mutations in thymidylate synthase, the target of its active metabolite 5-FdUMP (5-fluoro-deoxyuridinemonophosphate; see Chap. 18, Sec. 18.3.2). Similarly, resistance to methotrexate may occur because of the production of variant forms of DHFR, the target enzyme for this drug (see Fig. 19–1). Variant enzymes have been found that retain adequate function for reduction of their normal substrate (dihydrofolate) but have decreased affinity for methotrexate.
Another mechanism leading to methotrexate resistance is elevated production of DHFR resulting from an increase in the number of copies of the DHFR gene (gene amplification) (see Fig. 19–1; Schimke, 1984). High levels of methotrexate resistance in cultured cells is usually observed after stepwise increases in the drug concentration in the medium, and this may lead to as many as 100 to 1000 copies of the DHFR gene. Amplification of DHFR has also been observed in human lung tumors from patients treated with methotrexate.
Although gene amplification has been studied most extensively in relation to methotrexate resistance, there is increasing evidence for the importance of this mechanism in determining resistance to several other drugs, including upregulation of the target enzyme for the active metabolite of 5-FU (thymidylate synthetase) (Watson et al, 2010). Amplification of genes encoding 1 or more of the ABC drug efflux pumps (P-glycoprotein, MRP1, or ABCG2/BCRP), can also lead to multidrug resistance phenotypes (Cole et al, 1992; see Fig. 19–6), although amplification of these ABC genes is rarely detected in patient samples (see Sec. 19.2.3).
Drug resistance caused by gene amplification may be either stable or unstable when cells are grown in the absence of the drug. Stable amplification is typically associated with a chromosomal location of the amplified genes, often seen as homogeneously staining regions (HSRs) in stained chromosome preparations. Unstable amplification is usually associated with location of the genes in extrachromosomal chromatin structures known as double minutes (DMs). Multiple gene copies at both locations may be evident during selection for drug resistance (see Fig. 19–6).
19.2.5.2 Resistance to Drugs Targeting the DNA Topoisomerases DNA topoisomerases are nuclear enzymes that catalyze topological changes of DNA structure required for recombination and replication of DNA and for transcription of RNA. These enzymes also play a central role in chromosome structure, condensation/decondensation, and segregation (Nitiss, 2009; Pommier, 2009). Under physiological conditions, these covalent enzyme-DNA cleavage complexes are short-lived intermediates present at low concentration that are well tolerated by the cell. Topoisomerases serve as cellular targets for several important antineoplastic agents, some of which appear to stabilize the DNA-enzyme complex, leading to increased DNA strand cleavage, and thereby mediating, at least in part, the cytocidal activity of these compounds (see Chap. 18, Sec. 18.4). Other drugs targeted to topoisomerases act as conventional enzyme inhibitors and do not require that the topoisomerase be bound to DNA to be toxic (Nitiss, 2009; Pommier, 2009).
Camptothecin and its close structural analogs topotecan and CPT-11 exert their antitumor activity by inhibiting the 100-kDa topoisomerase I (see Chap. 18, Sec. 18.4.1). Under physiological conditions, the enzyme produces transient single-strand breaks in DNA and binds covalently to the 3’-phosphoryl end of DNA at the break site through a tyrosine residue at position 723 in the COOH-terminus. It then facilitates passage of an intact DNA strand through the break site, followed by religation of the cleaved DNA. Camptothecin (and its analogs) form a reversible complex with topoisomerase I and DNA, which shifts the equilibrium reaction markedly in the direction of cleavage. This results in increased DNA damage and, ultimately, cell death. Downregulation and production of mutant forms of topoisomerase I have been reported in cells resistant to these drugs (Pommier, 2009). Point mutations involving amino acid residues 361-364 appear particularly critical for resistance to camptothecin and its derivatives. Analyses of the topoisomerase I crystal structure indicate that this region is important for hydrogen bonding to camptothecin and it is close to the catalytic tyrosine residue and the bound DNA (Urasaki et al, 2001).
All vertebrates have 2 forms of topoisomerase II: an α-isoform (170 kDa), encoded by the human TOP2A gene on chromosome 17q21-22, and a β-isoform (180 kDa), encoded by the TOP2B gene on chromosome 3p24. Both enzymes function as homodimers and the mechanisms by which they alter the topology of double-stranded DNA, like topoisomerase I, require a catalytic tyrosine residue in each topoisomerase II subunit for binding to DNA. Distinct from topoisomerase I, the catalytic cycle of topoisomerase II is ATP-dependent.
Drugs that target topoisomerase II are divided into 2 broad classes (see Chap. 18, Sec. 18.4). One class includes the clinically important etoposide, doxorubicin, and mitoxantrone, and these drugs “convert” topoisomerase II into a DNA-damaging agent by increasing the levels of topoisomerase II/DNA covalent complexes and DNA double-stranded breaks (DSBs). The DSBs caused by drug-stabilized topoisomerase II-linked DNA, although often repaired by a complex array of nucleolytic and proteolytic pathways (see Chap. 5, Sec. 5.3), may lead to cell death. Decreases in topoisomerase IIα content or activity caused by alterations in TOP2A transcription or increased protein degradation, as well as point mutations and small deletions that alter the enzyme’s ability to bind DNA and/or drug, are associated with resistance to topoisomerase II poisons in numerous cultured cell lines. Mutations associated with resistance to the topoisomerase “poisons” cluster around the ATP-binding site or around the catalytic tyrosine residue in the DNA-binding region of topoisomerase II. The prevalence of these TOP2A mutations in clinical samples is not known, but several studies suggest it is low. Conversely, a good correlation with increased sensitivity to anthracyclines has been found in human tumors where TOP2A has been coamplified with the nearby ERBB2/NEU on chromosome 17 (Mano et al, 2007).
Although closely related in amino acid sequence homology (>70%), topoisomerase IIα and β differ in biochemical and biophysical characteristics as well as in their tissue-specific expression, subcellular localization, cell-cycle dependence, and sensitivity to some antineoplastic agents (Nitiss, 2009). Nevertheless, many drugs target both isoenzymes, which may have implications for reducing the risk of acquiring resistance because 2 targets are involved. Targeting the β-isoform is associated with induction of cardiotoxicity and secondary malignancies, and likely provides less benefit than targeting the α-isoform. This has prompted a search for topoisomerase IIα-specific drugs which might have greater antitumor activity and reduced toxicity.
19.2.5.3 Resistance to Agents Targeting Microtubules Microtubules are dynamic polymeric structures comprised of heterodimers of α- and β-tubulin (each with several subtypes) that are assembled and disassembled as required during many cellular events, including cell movement and intracellular transport, and are particularly important regulators of cell-cycle progression. Thus microtubules play a critical role in mitosis and drugs that bind to tubulin may cause mitotic arrest and cell death. These drugs may be classified according to where they bind: the Vinca alkaloids bind to the ends of microtubules, while taxanes bind along the interior surface of the microtubules (Dumontet and Jordan, 2010; see Chap. 18, Sec. 18.5). The mechanisms underlying resistance to the microtubuledestabilizing Vinca alkaloids vincristine and vinblastine, and the microtubule-stabilizing taxanes (eg, paclitaxel) are not fully understood. Mutations in βI-tubulin are found commonly in drug-resistant cell lines, but their clinical relevance appears questionable (Kavallaris, 2010). Abnormal high levels of expression of βIII-tubulin have been detected in drug-resistant solid tumors, including both lung and ovarian carcinomas. How elevated βIII-tubulin mediates resistance and enhances cell survival is complex, but this tubulin subunit helps to protect cells against the genotoxic stress induced by cytotoxic drugs. Microtubule-associated proteins have also been observed in drug-resistant cells. Microtubule-associated proteins (eg, tau) can bind to and stabilize microtubules against depolymerization. Binding of tau to the outer microtubule wall probably results in limited access of drugs, such as paclitaxel, to the inner luminal surface of the microtubule (Ferlini et al, 2007).
19.2.6 Drug Resistance and Repair of Drug-Mediated DNA Damage
DNA is constantly being subjected to damage by both exogenous and endogenous molecules. Many chemotherapeutic agents cause a variety of toxic DNA lesions that lead to cell death unless the damage is repaired. Detection and repair of drug-induced DNA lesions is carried out by lesion-specific DNA repair pathways (Helleday et al, 2008; see Chap. 5, Sec. 5.3). Resistance to DNA-damaging agents may occur because DNA repair processes in the tumor cells have become more efficient.
Cisplatin induces cell death by forming DNA-platinum adducts and interstrand DNA crosslinks but resistance can ensue if the lesions are repaired. Repair of platinum-modified DNA often involves nucleotide excision repair (NER; see Chap. 5, Sec. 5.3.3) and variations in NER activity can be an important determinant of drug sensitivity and resistance. For example, elevated levels of the excision repair cross-complementation group 1 (ERCC1) enzyme, which plays a rate-limiting role in the NER pathway, has been correlated with increased responsiveness to cisplatin-based adjuvant therapy in lung cancer patients (Olaussen et al, 2006). Conversely, defective NER has been implicated in the relative sensitivity of testicular cancer to cisplatin therapy.
Alkylating agents such as temozolomide also exert their cytotoxicity, at least in part, by binding to the guanine bases in DNA (see Chap. 18, Sec. 18.2.1). O6-methylguanine-DNA methyltransferase (MGMT) (also known as O6-alkylguanine DNA alkyltransferase) is one of the enzymes responsible for the repair of alkylated DNA. MGMT removes adducts from the O6 position of guanine and transfers the alkyl group to a specific cysteine residue (“acceptor site”) on the enzyme (Fig. 19–8). Because this transfer and alkylation of MGMT renders the enzyme inactive, the enzyme is considered to act by a “suicide” mechanism. Levels of MGMT or the methylation status of the MGMT promoter (which controls the expression of the gene) are useful predictors of the responsiveness of some tumors to alkylating agents (Esteller et al, 2000; Hegi et al, 2005). Potential opportunities exist to circumvent drug resistance mediated by MGMT through the use of O6-benzylguanine and related compounds. This relatively nontoxic agent can act as a noncompetitive substrate for MGMT, resulting in transfer of the benzyl group to the active site of the enzyme leading to irreversible inactivation (Fig. 19–8).
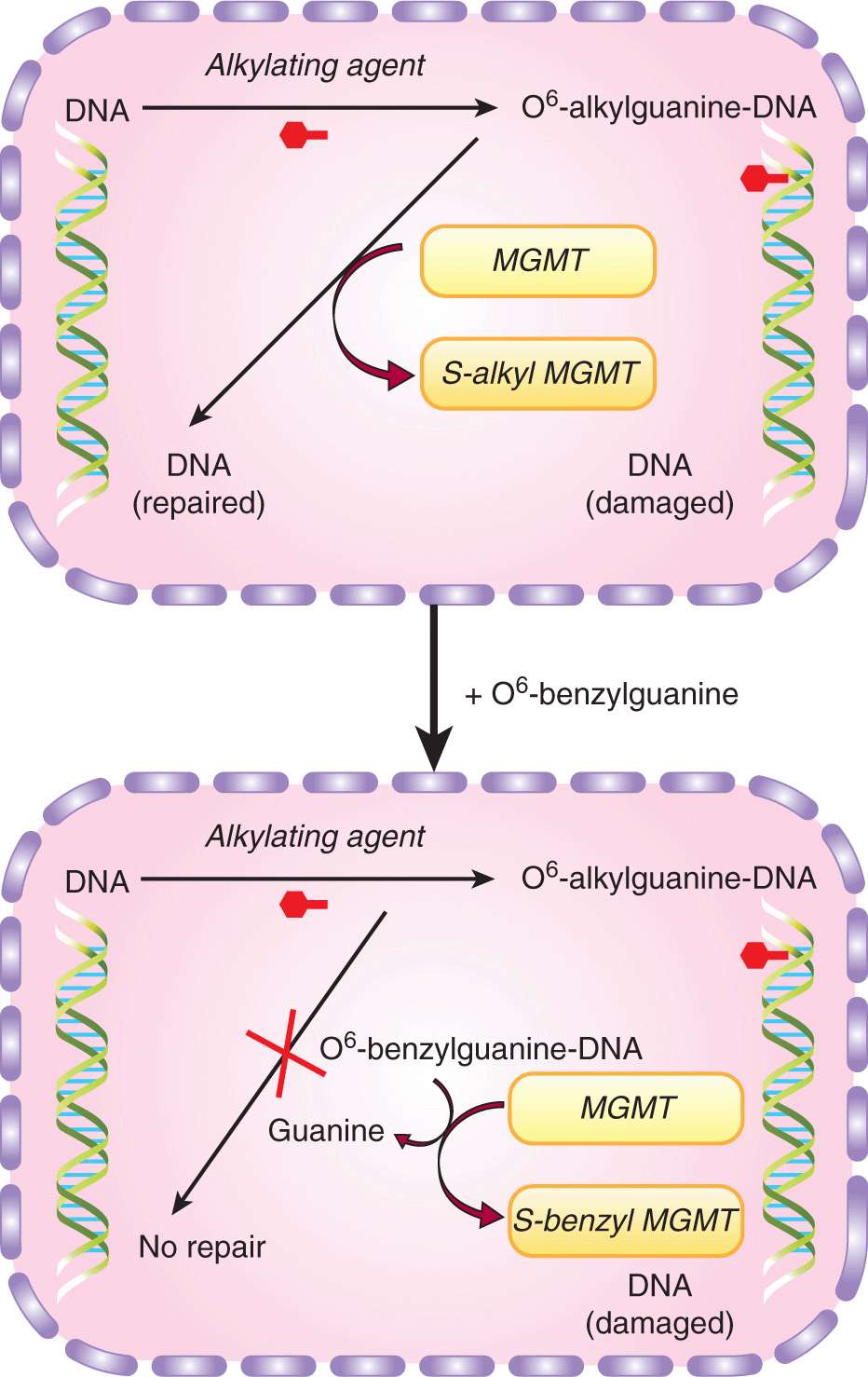
FIGURE 19–8 Resistance mediated by repair of an alkylated guanine base by MGMT and inhibition of this repair by O6-benzylguanine. Upper panel, the DNA repair enzyme MGMT causes resistance by removing toxic adducts from the O6 position of guanine in native DNA. It does this by transferring the alkyl group to a cysteine residue in the enzyme itself, resulting in auto-inactivation of enzyme; lower panel, in the presence of exogenous O6-benzylguanine, MGMT is no longer available to repair the DNA and resistance is circumvented.
Bifunctional alkylating agents, such as cisplatin, as well as drugs targeting topoisomerase I and II, can cause the accumulation of DSBs, leading to cell death unless the breaks are repaired. Repair of DSBs can take place by either nonhomologous or homology-directed repair pathways, the relative contribution of which depends on a variety of different factors (see Chap. 5, Sec. 5.3). Many human cancers probably have impairment in DNA repair pathways that contributes to their genomic instability, and such tumors are likely to be more sensitive to DNA-damaging agents. Regaining the capacity for certain types of DNA repair is a potential mechanism by which tumor cells can acquire resistance to DNA targeting drugs. For example, exposure to cisplatin of ovarian tumor cells containing BRCA2 mutations (which plays a crucial role in homologous recombination; see Chap. 5, Sec. 5.3.4) can select for drug-resistant cells, while additional mutations in BRCA2 can result in partial restoration of BRCA2 function and thus drug sensitivity (Sakai et al, 2008).
DNA damage caused by chemotherapeutic agents can cause a delay in cell-cycle progression in order to allow time for the damage to be repaired. Cell-cycle delays result from changes in the level, localization, or posttranslational modification of one or more of the proteins involved in the checkpoint processes that regulate specific phases of the cell cycle, and have been linked to drug resistance. Included among these proteins are complexes of cyclins and cyclin-dependent kinases (CDKs), which can be regulated by CDK inhibitors such as p21 and p27 (see Chap. 5, Sec. 5.4 and Chap. 9, Sec. 9.3.1). Because disruption of the cell-cycle checkpoints interferes with DNA repair by homologous recombination, modulating the activity of one or more of the proteins involved in checkpoint processes has been proposed as a means by which drug sensitivity can be increased. For example, absent or aberrant cytoplasmic localization of the G1-checkpoint proteins p21 (CDKN1A) and p27 (CDKN1B) has been linked to drug resistance in many in vitro cellular model systems (Abukhdeir and Park, 2009). Although convincing evidence of their relevance in clinical drug resistance is lacking, recent studies implicate both of them in resistance to the estrogen receptor modulator tamoxifen.
19.2.7 Resistance to Targeted Agents: The Tyrosine Kinase Inhibitors
The most widely used targeted drugs are small molecules, mostly derived from quinazoline, which act by inhibiting the kinase activity of oncogenes and/or growth factor receptors (see Chap. 17, Sec. 17.3.1). Other targeted agents include monoclonal antibodies with specificity for the external domain of cellular receptors, which thereby block binding of their endogenous ligands, and/or inhibit signaling from them. Thus agents that exploit mutations or amplification of the cKIT gene found in gastrointestinal stromal tumors, or the EGFR found in non-small cell lung cancers, or the human epidermal growth receptor2 (HER2/NEU) oncogene in breast cancer, or the BCR-ABL translocation protein product in chronic myelogenous leukemia (CML) have become important components in the therapy of these diseases (see Chap. 17, Secs. 17.3.1 and 17.3.3). Unfortunately, resistance to these drugs also occurs (Wheeler et al, 2010). For example, clinical resistance to imatinib, a small-molecule tyrosine kinase inhibitor, may result from point mutations that lead to diminished interaction of this drug with its target, the p210 BCR-ABL protein in CML (Milojkovic and Apperley, 2009). Resistance to imatinib in patients with CML is also associated with an increased copy number (amplification) of the BCR-ABL fusion gene (Gorre et al, 2001). Finally, imatinib is also a substrate of the P-glycoprotein and ABCG2/BCRP drug efflux pumps, which may limit the intracellular levels of imatinib that can be achieved (Dohse et al, 2010).
Response of lung cancer patients to the EGFR-targeted tyrosine kinase inhibitors erlotinib and gefitinib occurs in a subset of patients whose tumors have amplification of or mutations in the kinase domain of the EGFR gene (Gazdar, 2009). Subsequent studies led to the discovery of an EGFR mutation (T790M) that is associated with acquired resistance to these agents.
The limitations of imatinib and other small-molecule tyro-sine kinase inhibitors directed against a single membrane receptor prompted the development of receptor tyrosine kinase inhibitors (eg, sorafenib and sunitinib) that target multiple receptors. These compounds do not rely on active transport by SLC proteins to enter the cell nor are they efficiently transported by the major ABC drug efflux pumps (Hu et al, 2009). Consequently, these agents are likely to be less affected by transporter-mediated alterations in drug levels and transporter-related resistance mechanisms. These agents are useful in treatment of metastatic kidney cancer, but as for other anticancer drugs, resistance develops with continued use, through mechanisms not yet fully defined.
Mechanisms underlying resistance to monoclonal antibodies that target growth factor receptor kinases are poorly understood (Wheeler et al, 2010). For example, although no mutations in EGFR have been identified that predict response to cetuximab, KRAS mutation (at least in colorectal cancer patients) is an important predictive biomarker for response to this agent (Lievre et al, 2006). Also, among breast cancer patients whose tumors contain multiple copies of HER2 and respond initially to the monoclonal antibody trastuzumab, most experience disease progression within 1 year of initiation of treatment. The cause of resistance in the tumors of these women remains uncertain.
Resistance to all classes of tyrosine kinase inhibitors can also occur via activation of a parallel signaling pathway. For example, changes in the regulation of the signaling pathways downstream of the HER2 receptor have been implicated in trastuzumab resistance in breast cancer. Similarly, resistance to EGFR-targeted inhibitors may occur by a “bypass” mechanism mediated by amplification of the MET protooncogene, which, when activated by hepatocyte growth factor (HGF), increases tumor cell survival (Pal et al, 2010).
19.2.8 Resistance to Apoptosis and Autophagy
Most of the mechanisms of drug resistance described in previous sections are those in which the interaction of the anticancer drug with its target has been modified such that the activation of the pathways that normally leads to tumor cell death may be diminished. However, differences in the sensitivity of tumor cells to anticancer drugs also occur because of changes in the pathways that mediate cell death and survival. Types of cell death that have been observed following treatment with anticancer drugs include apoptosis, necrosis, mitotic catastrophe, and senescence, whereas autophagy may act (paradoxically) to enhance either death or survival of the cell. The best studied of these, apoptosis, appears to be initiated by the mitochondrial cytochrome c/Apaf-1/caspase-9 pathway (see Chap. 9, Sec. 9.4.3) while signaling through the death receptor pathway by drug-induced FasL upregulation seems less important, except possibly for 5-FU-induced cytotoxicity (McLornan et al, 2010). The proapoptotic and antiapoptotic members of the bcl-2 family, the kinases and phosphatases that regulate their activity and subcellular localization, the initiator and effector caspases, the various inhibitors of apoptosis proteins (IAPs), as well as the presence of wild-type or mutant p53, are all examples of proteins that might influence the sensitivity of tumor cells to apoptotic cell death (Brown and Attardi, 2005).
Laboratory studies provide evidence that in some cell types, drug sensitivity (as measured by a colony-forming assay or effects on tumor growth) can be modulated by changing the expression and/or posttranslational modifications of proteins that are components of apoptotic signaling pathways. For example, decreases in APAF-1 have been reported to contribute to therapeutic resistance of melanomas (Soengas et al, 2003). In addition, in a murine lymphoma, transfection of the bcl-2 gene led to increased resistance to multiple drugs (Schmitt et al, 2000), and the evidence for a contribution of blocked apoptosis to resistance of lymphomas/leukemias is quite convincing. However, several investigators have found little or no effect on drug sensitivity of solid tumors from modulating pathways of apoptosis (eg, Brown and Attardi, 2005). Markers of apoptosis such as an increase in TUNEL (terminal deoxynucleotidyl transferase-mediated deoxyuri-dine triphosphate-biotin nick-end labeling)-positive cells are observed commonly after treatment of malignant cells (or solid tumors) with anticancer drugs, but the clinical relevance of apoptotic pathways in determining drug sensitivity is not clear. Furthermore, studies of drug resistance in mice with conditionally mutated p53 and Brca1 as a model for hereditary breast cancer showed no changes in expression of Bcl2, Apaf1, or caspase genes after exposure to doxorubicin and paclitaxel. Rather, the major mechanism of resistance was upregulation of the P-glycoprotein drug efflux pump (see Sec. 19.2.3; Rottenberg et al, 2007). Similarly, exposure of these mice with Brca1-associated breast cancer to the topoisomerase I inhibitor topotecan resulted in drug resistance caused by reduced levels of the topoisomerase I drug target and increased expression of the Abcg2 drug efflux pump rather than changes in markers of apoptosis (Zander et al, 2010).
The p53 gene plays a role in apoptosis, and normal wildtype p53 can stimulate apoptosis of cells that have sustained damage to DNA (see Chap. 5, Sec. 5.3). A mutant p53 gene, present in a substantial proportion of human cancers, may inhibit apoptosis. The p53 protein may influence response to anticancer drugs, and modification of apoptosis is probably one of several mechanisms of drug resistance that can be influenced by it. For example, when wild-type p53 was induced in colon cancer cells expressing a mutant p53 protein, sensitivity to 5-FU, camptothecin, and radiation was increased (Yang et al, 1996). In another study, restoration of p53 function led to regression of lymphomas and sarcomas in mice (Ventura et al, 2007). Loss of p53 function was also reported to confer highlevel multidrug resistance in neuroblastoma cells (Xue et al, 2007). More recently, the chemosensitivity of human tumor cells, both in vitro and in mice, could be enhanced by using an adenovirus vector to increase p53 levels (and decrease p21 levels) (Idogawa et al, 2009). There is evidence that hypoxia in solid tumors can lead to a selective growth advantage for cells that express a mutant p53 gene, leading to a drug-resistant population (see Sec. 19.3.2 and Chap. 12, Sec. 12.2.2). Finally, p53-related transcription factors p63 and p73 appear also to be involved in a microRNA-dependent circuit that mediates inducible drug resistance (Ory et al, 2011).
A key determinant of whether the process of apoptosis might be modified to influence drug sensitivity is whether it is primary in causing lethal damage to cells, or simply represents a pathway whereby cells that have already sustained lethal and nonrepairable damage undergo cellular lysis (Fig. 19–9; Brown and Attardi, 2005). It appears likely that this depends on both the drugs used and the cell type that is treated; ongoing efforts to evaluate therapeutic strategies that target proapoptotic signaling pathways (see Chap. 9, Sec. 9.4.2) should help to distinguish between these possibilities.
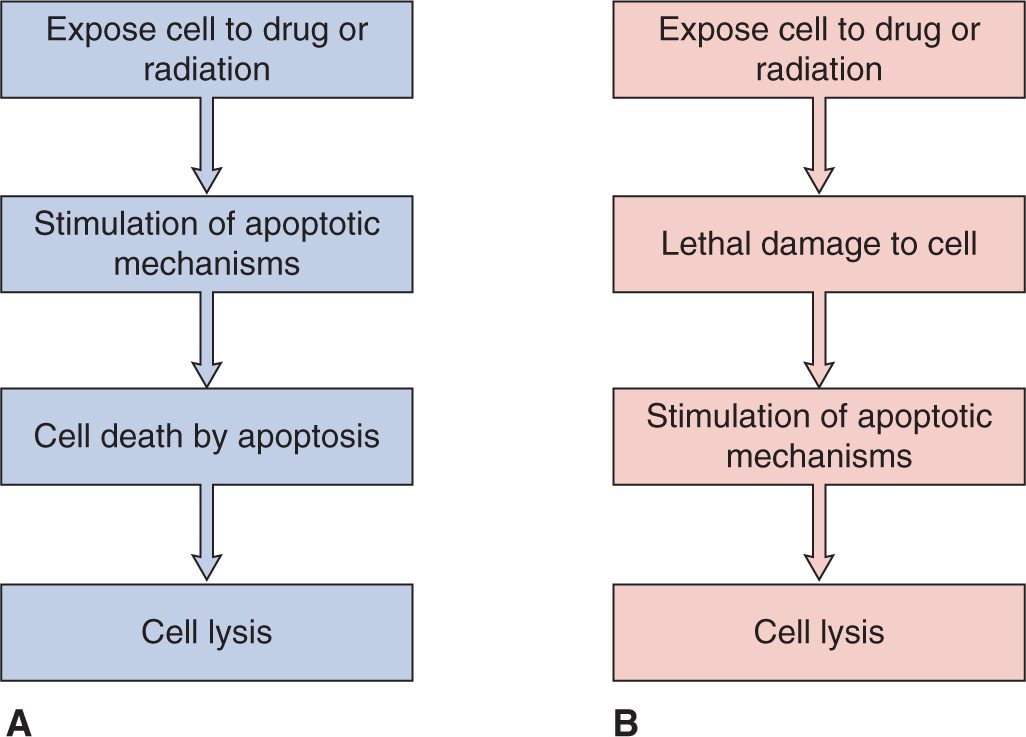
FIGURE 19–9 Apoptosis as a primary or secondary mechanism of cell lysis after drug treatment. (Adapted from Tannock and Lee, 2001.) signaling pathways (see Chap. 9, Sec. 9.4.2) should help to distinguish between these possibilities.
There is growing interest in the role of autophagy in tumor development and response to cancer therapy (see Chap. 12, Sec. 12.3.7). Autophagy is a lysosomal degradation pathway for intracellular digestion of cellular macromolecules that maintains cellular metabolism in times of stress such as exposure to hypoxia or to cytotoxic drugs. When autophagy is activated, intracellular membrane vesicles form and engulf proteins, cytoplasm, organelles and protein aggregates, and these vesicles are then delivered to lysosomes where they and their contents are degraded. Autophagy-associated pathways can promote cell survival by limiting damage and conferring stress tolerance, especially under adverse conditions (White and DiPaola, 2009). The use of inhibitors of autophagy could therefore be a promising strategy for overcoming resistance. Hydroxychloroquine (HCQ) and proton pump inhibitors have been identified as inhibitors of autophagy because of their ability to block lysosomal acidification and autophago-some degradation (Marino et al, 2010). Several clinical trials of HCQ in combination with cytotoxic (docetaxel, temozolomide) and targeted (an HDAC inhibitor, a vascular endothelial growth factor receptor [VEGFR] inhibitor, gefitinib) agents are underway, based on the premise that inhibition of autophagy by HCQ should enhance the efficacy of these drugs. Inhibitors of autophagy with greater specificity than HCQ are also being developed.
It is important to gain a better understanding of the relative contributions of the signaling pathways and modes of cell death that result in resistance to anticancer drugs. Establishing the clinical relevance of these potential mechanisms of drug resistance will be crucial before effective strategies for their reversal can be implemented.
19.3 DRUG RESISTANCE IN VIVO
Many laboratory-based studies of drug-resistance mechanisms have followed an approach whereby cultured tumor cells at low density are exposed to repeated selection in increasing concentrations of the anticancer drug of interest. These studies have led to the characterization of multiple mechanisms of drug resistance, described in previous sections, many of which are relevant to human cancer. However, the selection of stable drug-resistant subpopulations, present because of mutation or gene amplification, provides an oversimplified model for drug resistance in human cancer. Drug resistance of human tumors often occurs without prior drug exposure or may emerge after brief exposure to a relatively low concentration of drugs that is achievable in human tissue. In contrast, the selection of genetically stable drug-resistant mutant cells in experimental systems is often difficult, and typically requires much higher selection pressures (eg, prolonged exposure to mutagens, higher doses, and longer duration of exposure to anticancer drugs) than may occur during the treatment of human tumors. Also, when human tumors relapse after initial chemotherapy, they may respond later to the same chemotherapy (Cara and Tannock, 2001), suggesting that at least in some instances, the resistant phenotype may be transient.
Clinical drug resistance may occur as a result of mechanisms that depend on the in vivo microenvironment. Tumors have a complex extracellular matrix (see Chap. 10, Sec. 10.2), and cells in common epithelial-derived tumors have close cellular contact. Solid tumors have a poorly formed vasculature, which leads to regions of hypoxia and extracellular acidity (see Chap. 12, Sec. 12.2), and a requirement that anticancer drugs penetrate over relatively long intercapillary distances (as compared to those in normal tissues) to reach the target tumor cells (Minchinton and Tannock, 2006; Trédan et al, 2007; Fig. 19–10). Variable concentration of nutrient metabolites in the extracellular environment, and other factors, lead to variable rates of cell proliferation before treatment, and of repopulation of surviving tumor cells after treatment, both of which influence drug sensitivity of experimental and clinical tumors.
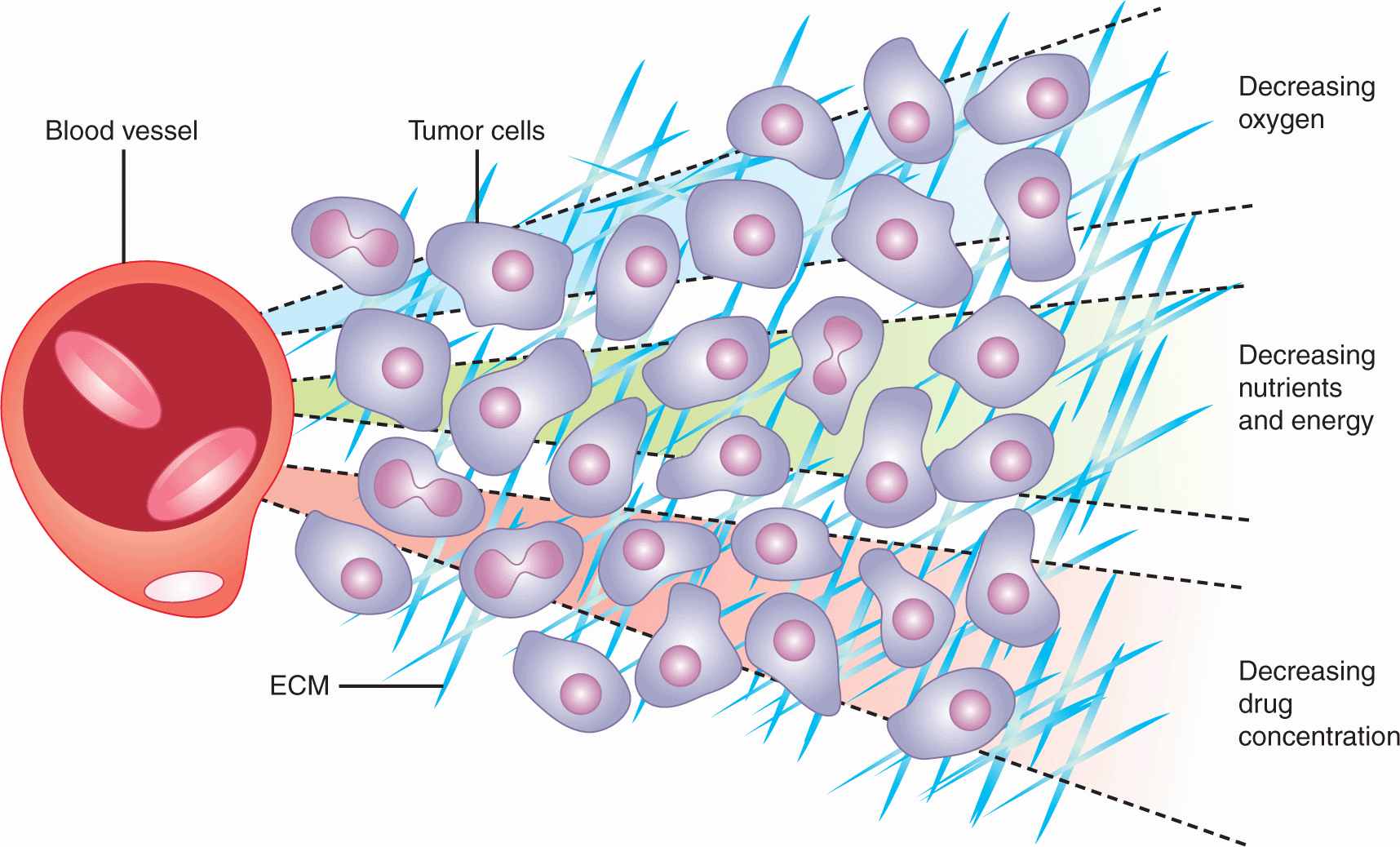
FIGURE 19–10 Representation of gradients of oxygen and other metabolites, and of cell proliferation and death in relation to blood vessels in solid tumors. ECM, extracellular matrix (Adapted from Minchinton and Tannock, 2006.)
Drug-resistance mechanisms that depend on the microenvironment may be explored by using model systems that maintain cellular interactions with other cells and with the extracellular matrix, thereby better reflecting how tumors in vivo are exposed to drugs. One model is provided by spheroids, in which malignant cells grow in contact with each other and with an extracellular matrix to form nodules in tissue culture (Durand, 1989; Hirschhaeuser et al, 2010; Fig. 19–11A). Alternatively, tumor cells can be grown on collagen-coated semiporous Teflon membranes to form multilayered cell cultures (MCCs) of relatively constant thickness to provide a useful model for studying drug penetration through tumor tissue (Hicks et al, 1997; Tannock et al, 2002; Fig. 19–11B; see Sec. 19.3.3). Drug can then be added on one side of the MCC and its time-dependent concentration measured on the other (Fig. 19–11C) to quantify the penetration of drugs through the MCC as compared to that through the semipermeable membrane alone (Fig. 19–11D). Such model systems have allowed study of mechanisms of drug resistance that depend on the cellular environment, as well as metabolic factors such as hypoxia and acidity. They supplement direct studies of the relationship between drug concentration, activity, and microenvironment in solid tumors that are either transplanted or induced in experimental animals (Trédan et al, 2007).
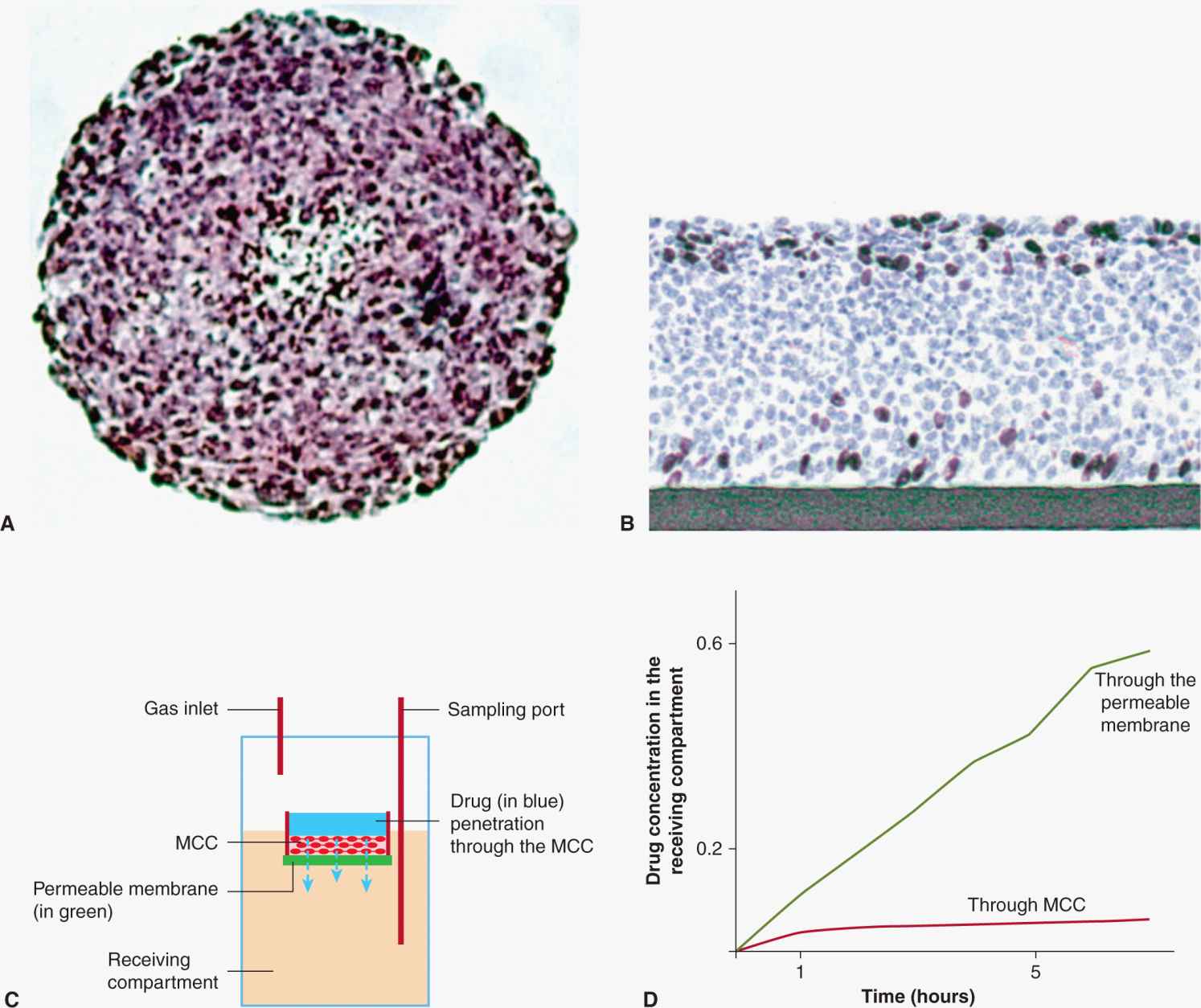
FIGURE 19–11 Models used for study of drug resistance that depends on the microenvironment found in solid tumors. A) Multicellular tumor spheroid. The distribution of fluorescent compounds may be imaged directly. B) Multilayered cell culture (MCC) grown on a collagen-coated Teflon membrane. C) The MCC is floated on medium in a larger vessel and drug can be added to the upper compartment in dilute agar and sampled as a function of time in the stirred lower compartment, on the other side of the MCC, to evaluate. D) The time-dependent transport of drug through the MCC as compared to that through the semiporous membrane alone. (From Trédan et al, 2007.)
19.3.1 Influence of Cell Contact and the Extracellular Matrix
Repeated drug treatment of solid tissue, either in the form of spheroids or tumor-bearing mice, may lead to drug resistance that is expressed only when the cells are grown in contact with one another. The tumor cells do not display drug resistance when grown without cell-cell contact as in dilute cell culture (Teicher et al, 1990; Kerbel et al, 1994). Further work suggests that drug resistance is correlated with the density of cell packing in spheroids, is dependent on integrin-mediated cell adhesion, and may be reversed by silencing of the gene encoding focal adhesion kinase (FAK), or by agents that inhibit adhesion between the cells (Chen et al, 2010; St Croix et al, 1996; Zutter, 2007; Chen et al, 2010). Integrin-mediated cell adhesion may influence multiple cellular properties, including cell proliferation, through regulation of cell-cycle checkpoints and upregu-lation of cell-cycle-inhibitory CDKs (Zutter, 2007; see Chap. 9, Sec. 9.2.2). Hence these effects may be mediated in part by a reduction in the rate of cell proliferation in the solid tissue environment, with consequent resistance to cycle-active drugs.
Many drugs have been tested against tumor cells in dilute tissue culture (eg, at a concentration of ~105 cells/mL) and there is an implicit assumption that relative cell kill will be similar at higher cell concentrations such as are found in solid tumors (108 to 109 cells/mL). This assumption of first-order kinetics is usually correct for drugs that are present in much higher concentration than their molecular targets, as is probably true for many anticancer drugs that kill cells by inducing a limited number of critical lesions in DNA. This assumption may not be correct for agents that must interact with a large number of cellular targets in order to be effective. For example, agents such as verapamil or cyclosporine, which reverse multidrug resistance caused by P-glycoprotein in dilute tissue culture, may lose their effect as the cell concentration in tissue culture increases (Tunggal et al, 1999); these agents have not been adequately evaluated against solid tumors in animals, and this effect may contribute to lack of efficacy of inhibitors of P-glycoprotein at cell concentrations that are observed in solid tumors (Yu et al, 2012). Inactivity at high cell concentrations could also be a problem for some other agents that are directed against targets on the cell surface, such as those that inhibit growth factor receptors, if there are a large number of targets per cell.
The drug sensitivity of tumor cells can be modulated by direct contact with other components of the tumor cell environment. Thus both in vitro and in vivo studies show that contact with the extracellular matrix can provide tumor cells with protection against cell death mediated by anticancer drugs, described as cell-adhesion-mediated drug resistance (CAMDR; Shain and Dalton, 2001). CAMDR has been most studied in multiple myeloma, where myeloma cells interact with stroma in the bone marrow, leading to initiation of survival signals that are absent when the same cells are in suspension (Li and Dalton, 2006). Several strategies to circumvent this type of resistance have been proposed that target molecules in tumor cells and/or in the extracellular matrix that are required for cell adhesion. The activity of the proteosome inhibitor bortezomib, which has led to improved prognosis in patients with multiple myeloma, appears to depend on its ability to inhibit CAMDR (Yanamandra et al, 2006; Noborio-Hatano et al, 2009).
19.3.2 Drug Resistance in Hypoxic Environments
Tumor vasculature is characterized by irregular blood flow and stasis, and by relatively large intercapillary distances in comparison to those in normal tissues (see Chap. 11, Sec. 11.5 and Chap. 12, Sec. 12.2). This leads to regions of tumors that are hypoxic, and, in turn, to regions where the extracellular pH is relatively low because of the production of lactic acid, and to poor clearance of this and other acidic products of metabolism (see Fig. 19–10). Hypoxia is widely recognized as a major factor leading to resistance of tumor cells to radiotherapy (see Chap. 16, Sec. 16.4), but several mechanisms may also cause cells in hypoxic regions to be resistant to anticancer drugs, as reviewed below.
Cells in nutrient-deprived regions of tumors tend to have a low rate of proliferation in comparison to cells situated close to functional blood vessels. Most anticancer drugs, including some molecular-targeted agents, are more toxic to proliferating than to nonproliferating cells (see Chap. 17, Sec. 17.5). Thus, even if the drugs achieve potentially cytotoxic concentrations in these regions, the level of cell kill may be limited. Surviving cells that were previously hypoxic may begin to proliferate following loss of killed cells closer to blood vessels, leading to improvement in the distribution of oxygen and nutrient metabolites, and therefore may allow regrowth of the tumor (see Sec. 19.3.4).
Hypoxia and extracellular acidity have direct effects on the activity and/or uptake of some anticancer drugs, independent of proliferative status. As for ionizing radiation, the toxicity of some drugs is dependent on the production of free radicals, and this process depends on availability of oxygen. Drugs that require active transport into cells are dependent on ATP, and anaerobic metabolism is much less efficient than oxidative phosphorylation in producing ATP. Drugs that are weak bases, such as doxorubicin, have a greater proportion of molecules in the charged form under acidic conditions, which decreases their ability to cross the plasma membrane and be taken up into the cell, leading to decreased activity. In contrast, extracellular acidity may enhance the uptake of drugs that are weak acids, such as chlorambucil or melphalan. In general, however, the direct effects of hypoxia and acidity on drug sensitivity are smaller than those on radiation sensitivity.
Hypoxia may influence genetically-based mechanisms of cellular drug resistance in at least 2 ways. Transient exposure to hypoxia, as may occur in tumors because of fluctuations in blood flow, may stimulate the amplification of genes, including those encoding DHFR, which leads to resistance to methotrexate (see Fig. 19–1; Rice et al, 1986; Sec. 19.2.5). Also, cells in many tumors do not express wild-type p53 (see Chap. 7, Sec. 7.6.1). Hypoxia has been found to provide a selective survival and growth advantage for cells lacking wild-type p53, because such cells show a diminished rate of apoptosis under hypoxic conditions (Graeber et al, 1996). This effect may help to explain why the presence of hypoxia in tumors is a poor prognostic factor after all types of management (including surgery). Following treatment with anticancer drugs that leads to selective killing of rapidly proliferating aerobic cells situated closer to tumor blood vessels, there may be selective repopulation of the tumor by p53(–/–) cells, which have survived hypoxic conditions, and which may be resistant to many therapeutic agents (see Sec. 19.2.8). The outgrowth of such p53(–/–) cells can confer resistance to treatment directed against tumor blood vessels, as well as to more conventional therapy (Yu et al, 2002).
Drugs are being developed that are selective for killing of hypoxic cells, and which might complement both radiotherapy and conventional chemotherapy. Hypoxia-activated prodrugs are inactive in their native form, but are activated by reduction under hypoxic conditions such as may occur in solid tumors, and may then kill tumor cells. The first agent to be tested clinically, tirapazamine, did not improve the activity of cisplatin and radiotherapy in randomized controlled trials, probably because it has poor diffusion characteristics into hypoxic regions. It also had toxicity to normal tissue (Rischin et al, 2010). Newer agents, such as PR-104 and TH-302, have more favorable properties and are under clinical development, although low levels of hypoxia in some normal tissues, including bone marrow, may lead to some normal-tissue toxicity.
19.3.3 Drug Access and Tumor Cell Resistance
Effective treatment of solid tumors requires both that the constituent cells be sensitive to the drug(s) that are used, and that the drugs achieve a sufficient concentration to exert lethal toxicity for all of the viable cells in the tumor. This depends on the efficient delivery of drugs through the vascular system of the tumor, and penetration of the drugs from tumor capillaries to reach tumor cells that are distant from them. Such cells will be in nutritionally deprived environments, as oxygen and other nutrients must gain access to the tumor by the same route, and may be relatively resistant to drugs for reasons described in the previous section (see Fig. 19–10).
The distribution of fluorescent or radiolabeled drugs in spheroids, and studies of their penetration through MCCs (see Fig. 19–11), indicate rather poor tissue penetration of multiple drugs, including doxorubicin, gemcitabine, taxanes, and methotrexate (Durand, 1989; Minchinton and Tannock, 2006). Smaller molecules distribute largely by diffusion, which depends on size, shape, charge, and solubility of the drug in the extracellular matrix, and “consumption” because of binding or metabolism by proximal cells. Larger molecules, such as therapeutic monoclonal antibodies, probably depend more on convection, which is inhibited by high levels of interstitial fluid pressure (Minchinton and Tannock, 2006). Both mechanisms depend on maintenance of a concentration gradient into tissue from tumor blood vessels, and distribution is therefore likely to be better for drugs with a prolonged lifetime within the circulation.
Drug distribution in relation to blood vessels in solid tumors can be quantified by using immunohistochemistry or autoradiography applied to tumor sections (Fig. 19–12). The blood vessels can be recognized by an antibody to an endothelial cell marker (eg, CD31), and patent vessels by an injected fluorescent marker, such as the carbocyanine dye DiOC7; hypoxic regions of tumors can be recognized by antibodies to injected markers of hypoxia such as pimonidazole or EF5. Drugs that are fluorescent, such as doxorubicin or mitoxantrone, can be recognized directly (Fig. 19–12A to C), while other agents (eg, monoclonal antibodies) can be recognized by fluorescent-tagged antibodies directed against them (Fig. 19–12D), or by autoradiography used to detect a radiolabeled form of the drug (as has been used to study the distribution of taxanes; Kuh et al, 1999). Pharmacodynamic markers of drug effect, such as changes in apoptosis (eg, activated caspase–3), cell proliferation (eg, Ki67) or DNA damage (eg, γH2AX) may also be tracked by immunohistochemistry using fluorescent antibodies that recognize these biomarkers. Computerized image analysis programs can relate drug concentration (eg, as measured by fluorescence of doxorubicin), or a marker of drug effect, with distance from the nearest blood vessel or nearest region of hypoxia over the whole area of a tumor section.
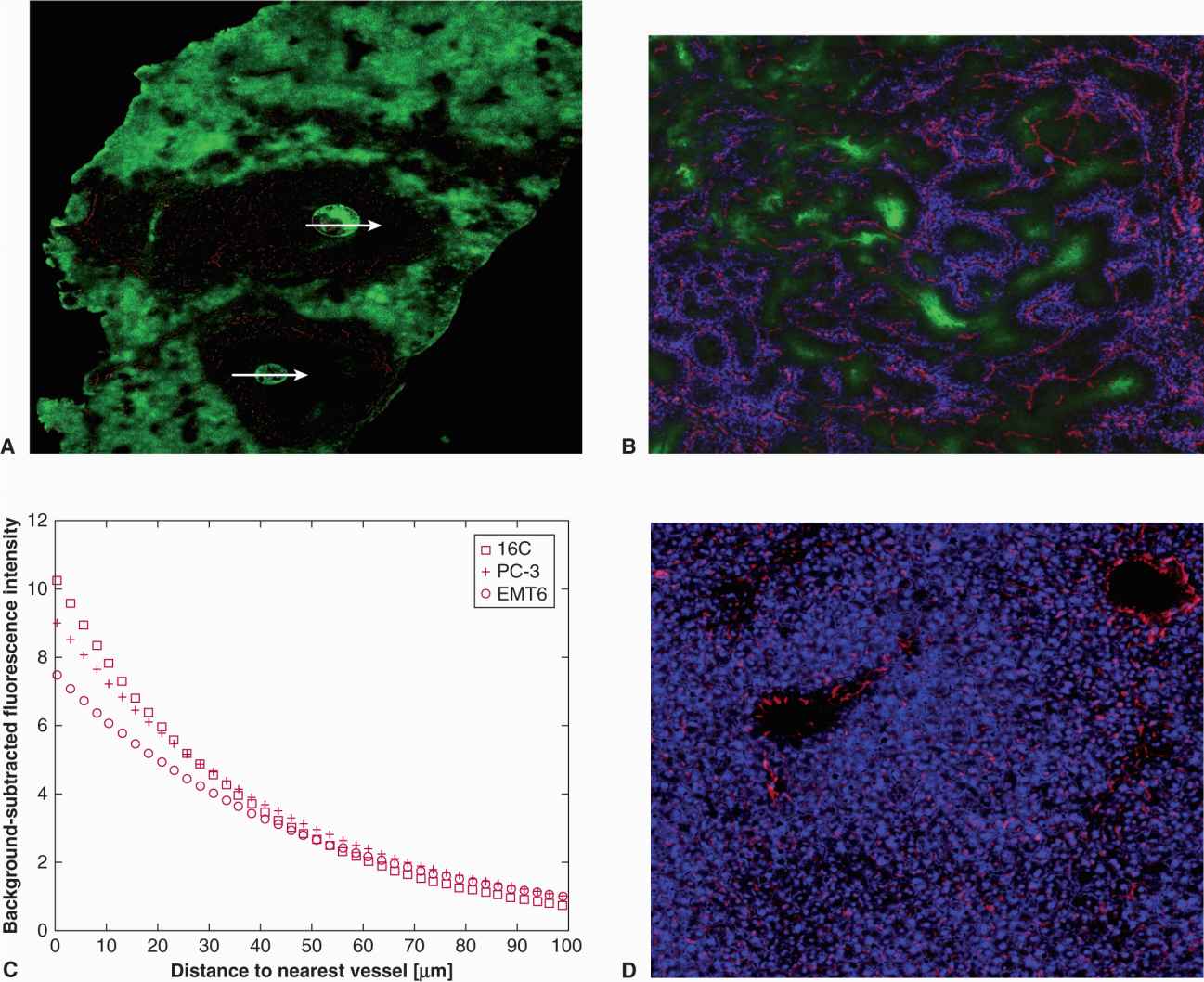
FIGURE 19–12 Drug distribution in tumor and normal tissue. A) Photomicrographs showing poor distribution of fluorescent mitoxantrone (green) into metastases (arrows) of a human breast carcinoma in the liver of a nude mice, as compared to normal liver. B) Distribution of doxorubicin (blue) in relation to blood vessels (red) and regions of hypoxia (green) in an experimental tumor. Note the perivascular distribution. C) Concentration of doxorubicin in relation to distance from blood vessels in 3 experimental tumors. D) Distribution of doxorubicin (blue) in relation to blood vessels (red) in normal mouse liver. (Parts B and C from Primeau et al, 2005.)
Studies using the above techniques show that for drugs that can be visualized directly by their fluorescence, there is a sharply decreasing gradient of drug from functional blood vessels of tumors (including human breast cancers) such that cells distal from blood vessels have minimal exposure after a single injection, with little or no penetration of such drugs into hypoxic tumor regions (see Fig. 19–12A and B; Lankelma et al, 1999; Primeau et al, 2005). This contrasts with more uniform distribution of drugs in most normal tissues, which have an orderly and functional vascular system (see Fig. 19–12C), although the presence of P-glycoprotein, MRP4, and other membrane transporters in the blood–brain barrier leads to uniformly low drug concentrations in brain. Similar poor drug distributions have been reported for taxanes and gemcitabine (Kuh et al, 1999; Huxham et al, 2004). Although the distribution of anticancer drugs in solid tumors might improve with sequential courses of chemotherapy, failure of drugs to penetrate tumor tissue in sufficient concentrations to kill cells distal from functional blood vessels is likely to be a major cause of poor clinical response.
Several strategies might be used to either modify or complement the distribution of anticancer drugs in solid tumors to improve therapeutic efficacy (Trédan et al, 2007). Agents that lower interstitial fluid pressure would be expected to improve distribution of drugs that is mediated in part by convection. Using another strategy, we have shown that administration to mice of proton pump inhibitors such as pantoprazole can improve the distribution of doxorubicin in solid tumors and increase its antitumor activity. This may occur because pantoprazole raises the pH of intracellular compartments (as it does in the stomach when used to treat ulcers) thereby displacing basic drugs, such as doxorubicin, that are sequestered within them, allowing more drug both to interact with target DNA and to distribute to cells more distal from blood vessels. However, proton pump inhibitors also inhibit autophagy (Marino et al, 2010; see Sec. 19.2.8 and Chap. 12, Sec. 12.3.7), which may be a survival mechanism for poorly nourished cells distal from blood vessels. Agents that modify the stromal cells or extracellular matrix may also improve drug distribution. For example, Tuveson et al have generated a genetically engineered mouse model of human pancreatic cancer that has dense stromal tissue, poor vascular perfusion, high interstitial fluid pressure, and resistance to chemotherapy, similar to human pancreatic cancer (Olive et al, 2009). Both IPI–926, a drug that inhibits the Hedgehog signaling pathway (see Chap. 8, Sec. 8.4.3), and hyaluronidase, an enzyme that targets the extracellular matrix, are reported to deplete the dense stroma and to improve the distribution and activity of anticancer drugs in such tumors (Olive et al, 2009; Provenzano et al, 2012); clinical trials evaluating such approaches are underway.
Hypoxia-activated prodrugs, described in the previous section, provide a potential method for complementing the action of conventional anticancer drugs that have limited penetration from tumor blood vessels, as they may act selectively against distal hypoxic cells that are exposed to low concentrations of the conventional agent. Once activated, some of these (hypoxia-dependent) drugs may also diffuse to kill cells in neighboring tumor regions.
19.3.4 Repopulation
Studies of the relationship between the probability of tumor control and duration of fractionated radiotherapy suggest that the proliferation of surviving tumor cells between daily doses of fractionated radiotherapy is an important cause of failure to achieve local tumor control (see Chap. 16, Sec. 16.6.2). Presumably this is a result of improving nutrition in the environment of surviving tumor cells as a result of killing of other cells in the tumor and better availability of nutrient metabolites and oxygen. For chemotherapy, the process of repopulation is likely to be more important because treatment courses are given typically at 3-week intervals, and in experimental studies a higher rate of proliferation (ie, of repopulation) has been reported after treatment of tumors by chemotherapy than in untreated control tumors (reviewed in Kim and Tannock, 2005). This process of tumor “recovery” may be analogous to repopulation of the bone marrow from stem cells that are stimulated to divide as a result of treatment. Few studies have assessed the rate of repopulation of human tumors following chemotherapy. This is problematic at early time intervals, because it is difficult or impossible to distinguish true surviving cells from lethally damaged cells that have yet to undergo the morphological changes that will precede their ultimate lysis, but less of a problem at longer intervals after drug treatment. There is evidence for accelerated repopulation and rapid regrowth of human tumors after completion of initial chemotherapy (Chen et al, 2011).
The possible effects of repopulation following chemotherapy have been modeled and are illustrated in Figure 19–13 (Kim and Tannock, 2005). If there is no change in the rate of repopulation, a human tumor may show net growth because of this process, even if each course of chemotherapy leads to killing of 70% of the viable tumor cells (Fig. 19–13A). However, a changing rate of repopulation can cause regrowth following initial tumor shrinkage (Fig. 19–13B), even if there is a delay in onset of repopulation after drug treatment caused by the immediate cytostatic effects of drugs on surviving cells (Fig. 19–13C). Tumor shrinkage or response followed by regrowth during continued treatment is observed commonly when chemotherapy is used to treat human tumors. The above modeling indicates that this can occur simply as a consequence of changes in the rate of repopulation of surviving cells after successive treatments and in the absence of any change in the intrinsic drug sensitivity of the constituent cells.
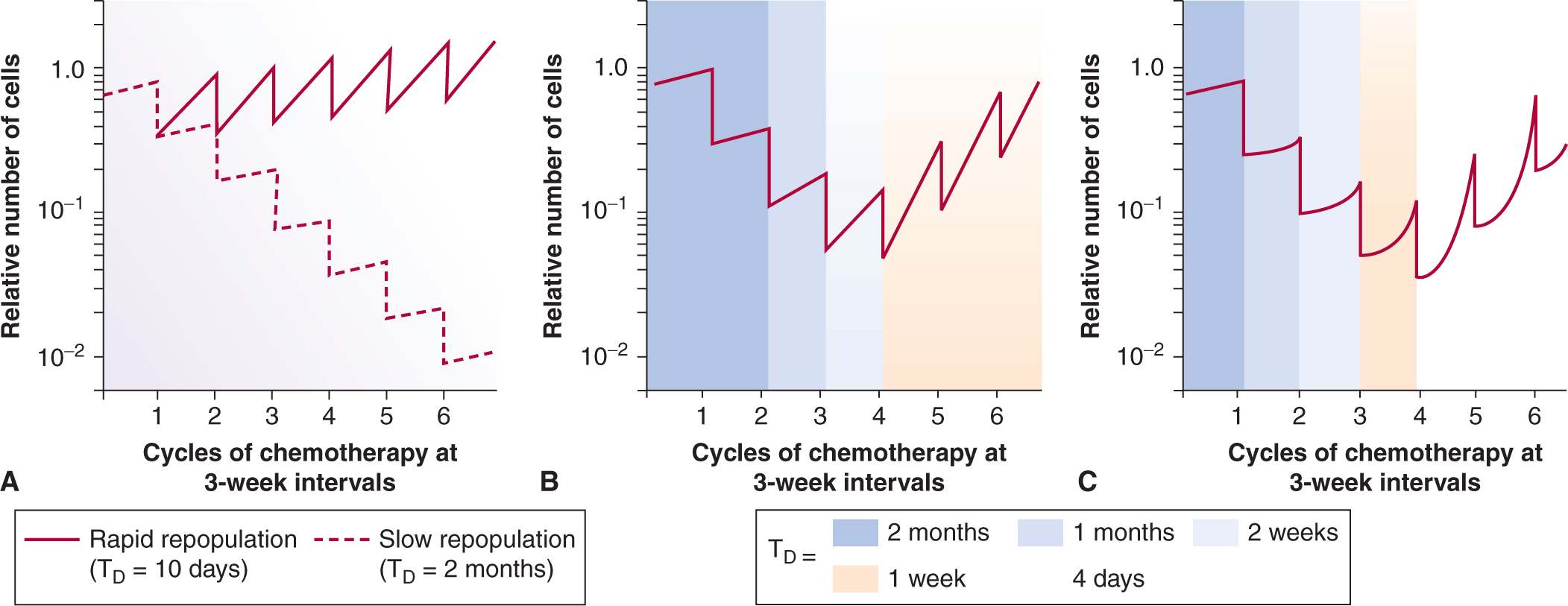
FIGURE 19–13 Models of cell killing and repopulation during chemotherapy. In (A) it is assumed that each 3-week course of treatment kills 70% of the tumor cells and repopulation is shown with a doubling time of 10 days (solid line) or 2 months (dashed line). In (B) the rate of repopulation increases between successive cycles of chemotherapy so that doubling time decreases from 2 months to 1 week. In (C) the model allows for initial cytostatic effects on surviving tumor cells after each dose of chemotherapy. Note that shrinkage and regrowth of tumors, observed commonly in the clinic, may occur as a result of accelerating repopulation and without any selection of drug-resistant cells. (From Davis and Tannock [2000].)
As for radiotherapy, there are strategies that might be used to inhibit repopulation between courses of chemotherapy, and thereby avoid the effective drug resistance that is observed. One method is to change the “fractionation” and to give lower doses of drugs more frequently or continuously. However, this is unlikely to be tumor specific, and may also lead to inhibition of cell proliferation in critical normal tissues such as bone marrow. Other approaches include inhibition of growth factor receptors that selectively stimulate tumor cell proliferation. Such agents will need to be short acting and discontinued just before the next cycle of chemotherapy, as anticancer drugs are likely to be more effective in killing cycling tumor cells.
19.3.5 Tumor Stem Cells and Drug Resistance
As described in Chapter 13, there is evidence that human tumors contain a small subpopulation of cells that have high potential to regenerate the tumor if they survive treatment, the putative human tumor stem cells, which are thus the important targets of treatment that aims to eradicate the tumor. Identification of specific proteins (markers) expressed on the cell surface have allowed for enrichment of this population in various types of tumors (eg, by using fluorescence-activated cell sorting after exposure to fluorescent-tagged monoclonal antibodies that recognize these markers), but it remains uncertain whether these markers truly characterize a stable population of stem cells, and how heterogeneity in their expression exists between cells within a tumor and between tumors. Studying the drug sensitivity of cells that bear stem cell markers is difficult because of their rarity within the tumor population and the instability of their phenotype in cell culture. However, there is emerging evidence that such cells may be more resistant to chemotherapy than other cell populations in the tumor and therefore may selectively survive during drug treatment (Dean et al, 2005; Eyler and Rich, 2008). For example, treatment of human breast cancer with initial (neoadjuvant) chemotherapy is reported to lead to a population of surviving cells with an increased probability of expressing stem cell markers, indicating their relative resistance compared to other tumor cells in the population (Li et al, 2008; Tanei et al, 2009).
Automated high-throughput screens have been devised to select for agents that might have specific activity against human tumor stem cells; for example, salinomycin was reported to be much more active than clinically used paclitaxel against cultured human breast cancer cells that were induced to undergo epithelial-to-mesenchymal transition, a process that is associated with acquiring stem-like properties (see Chap. 13, Sec. 13.3). However, it is uncertain whether this type of model is relevant to treatment of stem-like cells in human cancer, and investigation of drug screens that may reflect more directly the sensitivity of human stem cells is underway.
The relative drug resistance of such stem cell populations results from a variety of different mechanisms, including an enhanced capacity for DNA damage repair and changes in key molecules involved in oncogenic signaling (Maugeri-Sacca et al, 2011). Differential expression of several ABC transporters also has been widely observed in stem cell enriched-cell populations from a variety of tumor types, with ABCG2 being the most frequently detected of these (see Sec. 19.2.3). Elevated levels of P-glycoprotein (ABCB1) have been detected in stemlike cells compared with non–stem-like cells in ovarian cancer cell lines and in primary patient ascites (Rizzo et al, 2011) but in this study, the histone methyltransferase EZH2 appears to play a more important role in determining chemoresistance.
The sensitivity of tumor stem cells within human tumors is also likely to depend on their proliferative state and on their microenvironment within solid tumors (Jinushi et al, 2011). Cancer stem cells may have a relatively low rate of cell proliferation, like stem cells in normal human bone marrow (see Chap. 12, Sec. 12.1.3); their proliferative state will determine, in part, their sensitivity to cell-cycle active drugs. The location of stem cells in relation to tumor blood vessels will determine the probable concentration of anticancer drugs that can be achieved within their microenvironment (see Sec. 19.3.3). There is evidence that stem cells may exist in perivascular niches in some types of cancer (eg, brain tumors; Calabrese et al, 2007), whereas other evidence suggests that stem cells may be enriched in hypoxic regions of tumors, and relatively protected from drug access (Mohyeldin et al, 2010). In such cases hypoxia-activated prodrugs might play an important role in eradication of tumor stem cells (see Sec. 19.3.2).
Related posts:
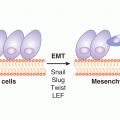
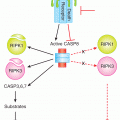
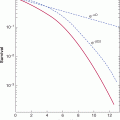
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


