Drug Resistance
Carl E. Freter
James Goldie
The capacity of cancer cell populations to express high orders of resistance to all of the currently available cancer therapeutic agents remains a formidable obstacle to more effective treatment for cancer patients. A relatively small subset of the total varieties of cancer to which human beings are prone can be cured by intensive courses of drug treatment. However, even in these instances, there remains the real possibility of relapse of the disease, generally in a state that is refractory to effective drug treatment.
The newer agents that have been introduced in the last few years (monoclonal antibodies, tyrosine kinase inhibitors, etc.), while expanding the utility spectrum of our therapeutic programs are still subject to the problem of resistance.
The ability of living systems to produce variant phenotypes that may be better adapted to certain environments appears to be a general property of all forms of life. This underlies the capacity of pathogenic organisms to alter their sensitivity to antimicrobial agents and indeed plays a critical role for the evolution of life on earth.
The first drugs that had a consistent therapeutic effect on at least some forms of disseminated cancer were introduced into clinical practice shortly after the end of World War II.1 Even as sometimes dramatic clinical remissions were observed, it was noted the patients’ tumors almost invariably recurred and became unresponsive to repeat applications of the initial chemotherapy.
Antibiotics were coming into widespread use at this time, and the similarities with acquired resistance in microbial populations suggested themselves. These parallels were reinforced by the studies of Law,2 which demonstrated that resistance to the folic acid antagonist methotrexate had the same biologic features that Luria and Delbrück3 had noted for bacterial resistance—that is, the drug-resistant variants appeared randomly within the tumor cell population consistent with a mutational origin. During the 1950s, a number of investigators were able to demonstrate biochemical changes in drug-resistant tumor cells that were consistent with the cells having acquired a resistant phenotype.4,5 For example, some resistant cells were shown to have impaired transport of the drug across the cell membrane or modification of the putative intracellular targets for the drug.6
Recent studies have shown a number of very broad mechanisms of resistance and, moreover, some of the molecular events that produce drug resistance are involved in the generation of the neoplastic state itself.7,8
In normal cell renewal systems, such as the bone marrow, there is a delicate balance between the proliferation (birth signals) and the pathways, leading to differentiation and apoptosis (death signals). Most of the cytotoxic agents appear to act through a final common pathway in which the damaged cell undergoes dissolution. In normal systems, the process acts very efficiently to cull abnormal cells from the population. In the malignant state, however, the death signaling machinery is impaired, permitting “unfit” cancer cells to survive. Although a few types of cancer retain a significant potential for undergoing apoptosis (i.e., lymphomas and germ cell tumors), most forms of cancer are not easily “pushed” into programmed or unprogrammed cell death. This clearly must be a major factor in determining so-called intrinsic resistance. In their review of this topic, Hahn and Weinberg8 indicate that there are three essential processes that must be activated to convert a normal human cell into a malignant one. These are (1) production of a continuous positive growth signal, (2) impairment of one or other of the pathways leading to apoptosis, and (3) activation of telomerase resulting in an indefinite life span for cells that would normally become senescent and die. Powerful oncogenic viruses such as the human papilloma virus illustrate this well.9 The virus’s genes code for proteins that, among other things, inactivate the proteins from the retinoblastoma gene and the p53 gene. These are two of the strongest proapoptotic signals in the cell, and the virus is exquisitely set up to inactivate them.
DEFINITIONS
The terms resistance and sensitivity are essentially relational statements and therefore, strictly speaking, should be defined in terms of some standard reference. In experimental, and particularly in in vitro, systems, this is relatively easy to establish, with the usual reference system being the parent cell line from which the resistant strain is developed. One can then pick an arbitrary concentration of drug and compare the amount of growth inhibition of the variant cell line with that of the parental line. Very simply, if growth were less inhibited in the variant cell line than in the parent, then we would define this as representing resistance. The resistance displayed by the variant cells is rarely absolute, and cells that are not inhibited by one concentration of a drug may well be inhibited by higher concentrations.
Cells that exhibit permanent drug resistance (i.e., show reduced growth inhibition essentially indefinitely) must be distinguished from cells that are temporarily less responsive to chemotherapeutic effect but may have their sensitivity restored under the right conditions and stimuli.
Distinction is also frequently made between so-called acquired drug resistance, which is the process whereby a tumor system
progressively changes its degree of sensitivity to a particular drug, and intrinsic resistance, which implies that the cell line displays a high order of drug resistance from the beginning, without having initially undergone selection by the drug in question.
progressively changes its degree of sensitivity to a particular drug, and intrinsic resistance, which implies that the cell line displays a high order of drug resistance from the beginning, without having initially undergone selection by the drug in question.
With in vivo tumors, either clinical or experimental, the definition of resistance is usually operational—that is, if a tumor fails to regress or progressively increases in size between treatment cycles of a drug given in the usual therapeutic dose, the tumor can be considered resistant. This need not mean that none of the tumor cells are being killed but simply that the number killed is lower than the net increase in cell number due to tumor growth, with the result that there is progressive increase in tumor burden over time. If the behavior of these treated tumors is compared with that of untreated controls, however, some overall reduction in tumor doubling time may be noted, with some extension in average survival. Strictly speaking, any displacement of the untreated growth curve toward some degree of retardation implies less than absolute resistance. On a practical basis, though, small shifts in net growth rate of the tumor are unlikely to translate into clinically useful outcomes.
BIOLOGIC BASIS OF DRUG RESISTANCE
As mentioned, there appear to be clear parallels between the development of resistance in tumor cell lines and the development of resistance to antibiotics by bacteria and other microbial species. Moreover, there are analogies to the phenomenon of intrinsic resistance in bacterial populations because different types of bacteria show variable degrees of inherent sensitivity to different antibiotics. For instance, Mycobacterium tuberculosis is poorly sensitive to penicillin and sulfanilamide but shows significant sensitivity to streptomycin and rifampicin.10 Even in the microbial systems, uniformly high sensitivity to all of the available antibiotics is not observed.
The origin of drug-resistant bacteria was the subject of considerable debate until the experimental studies of Luria and Delbrück3 in 1943 demonstrated that acquired resistance in bacteria was compatible with a mutational origin for the resistant phenotypes (Fig. 3.1). The fluctuation test of Luria and Delbrück used a statistical analysis of the distribution of resistant mutants in a bacterial population to distinguish between induction of or selection for resistant organisms. Application of this technique shows that agents such as antibiotics functioned in an essentially passive mode by eradicating the sensitive variants, leaving behind a resistant subpopulation. The work of Law referred to earlier2 showed that essentially the same process was occurring in transplanted mouse lymphoma cells with respect to their capacity to develop resistance to methotrexate.
The statistical technique used in the fluctuation test is called the analysis of variance. It uses a property of the Poisson distribution to compare the difference between two types of random processes.
If the resistant cells (or bacteria) are directly caused by exposure to the toxic agent, this will produce a single Poisson event in time. A property of the Poisson distribution is that the variance of the process (i.e., the square of the standard deviation) is equal to the mean of the distribution—that is, there is little variation from one induced event to the other. If the toxic agent is acting to select preexisting resistant cells from the population, however, the statistical distribution is different.
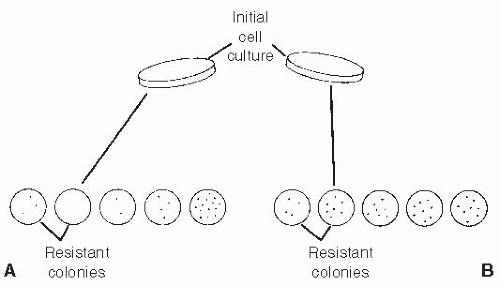 Figure 3.1 The Luria-Delbrück fluctuation test demonstrates the spontaneous nature of drug-resistant variant cells. In (A), parallel subclonal populations of cells are grown up to a predetermined number. These are derived from a parent stock culture. The individual cultures are then exposed to a cytotoxic agent. A very substantial variation, or fluctuation, in numbers of resistant cells is noted per individual culture tube, with the variance among the tubes being significantly greater than the mean for the whole test population. When similar-size samples of cells are drawn directly from a stock culture, put in a parallel series of tubes, and then immediately treated with the cytotoxic agent, much less individual variation in resistance cell colonies is seen from one culture to the other. The difference in the two sets of experimental results is related to the random and spontaneous nature of mutational events. In any one individual, subclonal culture displayed in (A), there is a probability that a mutation may occur early in the growth of the clone, producing a large number of resistant colonies. Likewise, there is also a probability that no resistant colonies may have emerged in a specific individual clone. Because the cultures depicted in (B) are simply aliquots taken from a large representative population, little variation in the numbers of resistant cells from culture to culture is seen, and little would be expected. A positive result in a fluctuation test such as this is consistent with the spontaneous origin of the variant cells, as opposed to their direct induction by the toxic agent. |
The preexisting resistant cells (i.e., mutants) can all be considered to have arisen from a series of mini Poisson events. The resistant cells present at the time of drug exposure will be the descendants of the cells produced by the prior series of Poisson events (mutations). When we have a number of Poisson events, the variance of each event is added, producing a much greater variance (much larger than the mean) than if there was just a single event, as in the case of a “directed” mutation. This will translate into a much greater variation in the numbers of resistant cells from one subclonal sample to another compared with the smaller variation seen in a directed mutation scenario. The point is labored here because the lessons of the fluctuation test still seem to be poorly understood—that is, drugs do not cause resistance to themselves to occur, but rather act on preexisting variation in the target population.
Some studies have used the fluctuation analysis to determine whether resistance to the commonly used agents etoposide and
doxorubicin arises through mutational or inducible events. Jaffrezou et al.11 found that in a line of human sarcoma cells, resistance to etoposide arose with a mutation rate ranging from 2.9 × 10-6 to 5.7 × 10-7. The lowest concentration of etoposide was associated with the highest value for the mutation rate. The clones derived from the highest value reverted to the sensitive mode. The other clones were stably resistant, and statistical analysis indicated that they had arisen by spontaneous mutation. In these cells, resistance was mediated by decreased expression of the target enzyme topoisomerase II.
doxorubicin arises through mutational or inducible events. Jaffrezou et al.11 found that in a line of human sarcoma cells, resistance to etoposide arose with a mutation rate ranging from 2.9 × 10-6 to 5.7 × 10-7. The lowest concentration of etoposide was associated with the highest value for the mutation rate. The clones derived from the highest value reverted to the sensitive mode. The other clones were stably resistant, and statistical analysis indicated that they had arisen by spontaneous mutation. In these cells, resistance was mediated by decreased expression of the target enzyme topoisomerase II.
Chen et al.,12 using the same line of human sarcoma cells, found that doxorubicin resistance arose with a frequency of 1.8 × 10-6 per cell per generation. Analysis indicated that these clones had arisen from spontaneous mutation. The mechanism of resistance in these cell lines appeared to be overexpression of the mdr-1 gene (see the section Multidrug-Resistant Phenotype).
Other types of evidence are also strongly consistent with a genetic and mutational origin of the resistant phenotype.13,14 These include studies in which a drug-sensitive and drugresistant cell are fused together, and the pattern of drug sensitivity in the resultant hybrid cell is then examined. In these situations, the hybrid cells display patterns of drug resistance consistent with those predicted from Mendelian laws of genetic inheritance. Even more direct evidence can be obtained from DNA transfection studies in which DNA segments coding for specific genes can be transferred from a drug-sensitive one, conferring permanent and heritable drug resistance on the transfected cell.15
Evidence suggests that chemotherapeutic agents commonly express their cell-killing effect by triggering apoptosis.16,17,18,19 Because apoptosis can be seen as one of the ways that cells prevent the accumulation of mutated and abnormal progeny, agents that produce genotoxic damage could be expected to be potent initiators of the process. It also implies that mutations affecting the apoptotic machinery could constitute another whole level where drug-resistance changes might manifest themselves. A growing amount of data suggests that this indeed is the case. Studies have suggested20 that renal cell carcinoma, one of the most obstinately drug-resistant forms of cancer, has mutations affecting the caspase system, the complex enzyme cascade that plays a key role in generating apoptosis.21
RESISTANCE DUE TO CELL KINETIC PHENOMENA
In addition to types of unambiguous genetic resistance, cells can exhibit reduced drug sensitivity by virtue of their position in the cellular division cycle. In the case of drugs that act primarily at one point in the cell cycle (e.g., during DNA synthesis), a cell may be quite invulnerable to the drug’s effect if it does not pass through S phase during the period of drug exposure. Moreover, cells that are temporarily out of the division cycle in a so-called resting state, or G0 state, may display significantly reduced drug sensitivity to a variety of agents (Fig. 3.2).
In these examples of so-called kinetic resistance, however, the phenomenon is generally only temporary. If the drug concentration is maintained long enough, all of the cells in the tumor may pass through the vulnerable point in the cell cycle and become susceptible to the drug’s effect. Likewise, cells that are in a resting state and are stimulated into division will become sensitive to drug effect.
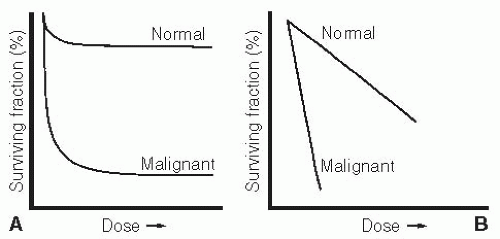 Figure 3.2 The two principal types of dose-response curves for cytotoxic agents measured in vivo. A: Demonstrates so-called phase-specific or cell cycle stage-specific classes of agents. These include agents such as methotrexate, vincristine, and cytosine arabinoside. Such drugs exert their killing effects primarily during one phase of the cell cycle, so cells that do not pass through this particular vulnerable point are not killed during relatively short exposure. The normal cells of interest here are the colony-forming cells of the normal bone marrow. The fact that a significantly lower proportion of these cells are killed for any particular dose of drug (compared with rapidly proliferating lymphoma cells) indicates that a much lower proportion of hemopoietic cells are in cell cycle during the period of exposure. B: Indicates a typical log-linear dose-response effect that is seen with alkylating agents and DNA intercalators. Increases in dose produce a linear increase in log kill of both normal and malignant cells, but again, there is a preferential killing effect on the malignant cells because a much higher percentage of these are in cell cycle at any given time. (Adapted from Bruce WR, Meaker BE, Valeriote FA. Comparison of the sensitivity of normal hematopoietic and transplanted lymphoma colony-forming cells to chemotherapeutic agents administered in vivo. J Natl Cancer Inst. 1966;37:233-245, with permission.) |
That is, they will be sensitive if they are inherently sensitive to begin with. If a genetically drug-resistant cell switches from a resting to a proliferative phase, it will not become drug responsive. In the studies of Bruce et al.,22 described in Figure 3.2, it is important to note that the switch between a resting and proliferative state with associated changes in drug sensitivity is a phenomenon that was observed in the normal, as opposed to the malignant, cells in the test system. Under steady-state conditions, most of the hematopoietic stem cells in the mouse marrow were in a physiologic G0 condition, with just enough stem cells entering the division cycle to replenish the differentiated cells lost through normal attrition. After perturbation by the chemotherapeutic agent, the surviving marrow stem cells all enter the generation cycle to replenish the marrow stem pool rapidly. Once this pool has regained its physiologic size, further expansion of the normal stem cells is inhibited through a feedback process. The tumor cells in this system showed no such physiologic responses to external stimuli. Virtually all of the malignant stem cells appear to be in the generation cycle, so their drug sensitivity did not change because of kinetic alterations. One therefore has to exercise caution in extrapolating the behavior of resting normal cells to what may or may not occur in neoplastic systems.
There is certainly evidence that malignant cells can enter a nondividing state from which they can be recalled into division by the appropriate stimuli.23 It is not clear from a mechanistic point of view, however, if this is analogous to the physiologic G0 state seen with the marrow stem cells. Indeed, if failure of so-called checkpoint arrest is one of the fundamental changes that occurs in malignancy, the concept of the G0 tumor cell needs to be questioned. Tumor cells may cease dividing under conditions in which nutrients and growth factors are limiting, but in vitro at least, this is the result of cells stopping at random points in the cell cycle.24
EXPERIMENTAL MODELS OF DRUG RESISTANCE
It is relatively easy to produce drug-resistant variant cell lines with established in vitro cell culture systems. A common method is to grow the cells in progressively increasing sublethal concentrations of a particular drug. When first exposed to the cytotoxic agent, the growth of the cell line is markedly inhibited, but over time, the cells appear to become adapted to the cytotoxic agent and start growing at a rate close to their initial pretreatment doubling time. At this point, the concentration of the agents in the culture media can be increased and then the process repeats itself. Depending on the cell line, the particular anticancer agent that is being used, and the degree of drug resistance that is desired, it generally takes from a few weeks to several months to produce a cell line that will now grow unaffected by concentrations of drug that initially would have been highly toxic (Fig. 3.3).
If the drug is now removed from the tissue culture environment, in most circumstances, the cell line will maintain its resistance without continuous drug exposure, although one may see a gradual reduction in drug resistance over time.
Such methods are very useful for obtaining stable cell culture lines that display high orders of drug resistance. This makes it feasible to carry out biochemical characterization studies of the resistant cells. It can be argued that this process of producing drug-resistant cells differs appreciably from what occurs in in vivo drug selection and that the high orders of drug resistance that are so developed may not be completely relevant to clinical situations.
If one uses an in vivo transplanted tumor as the experimental system, the usual approach is to inoculate a known burden of tumor cells into a host animal and then treat with a noncurative schedule of chemotherapy.25 As with the in vitro systems, these resistant cells generally maintain their degree of resistance over many passages in recipient animals. Some decline in resistance in the absence of periodic reselection may occur, however.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


