- Many commonly used drugs interfere with glucose homeostasis and can provoke hyperglycemia in subjects without diabetes or worsen glycemic control in patients with diabetes.
- Glucocorticoids act on post insulin-receptor mechanisms to induce dose-dependent insensitivity to insulin in its target tissues.
- Glucocorticoid-induced diabetes often requires treatment with oral hypoglycemic agents or insulin while glucocorticoid treatment continues.
- Oral contraceptive pills rarely cause hyperglycemia, although it can occur with combined pills that contain high-dose estrogen or the progesterone levonorgestrel, and in women with a history of gestational diabetes.
- Estrogen replacement therapy in postmenopausal women does not affect glycemic control.
- Thiazide diuretics impair insulin secretion when given at high dosages. Impairment of insulin secretion is related to potassium depletion.
- Loop diuretics such as furosemide (frusemide) are less likely to cause hyperglycemia.
- Non-selective β-adrenoceptor antagonists inhibit insulin secretion and can impair glucose tolerance. Cardioselective β1-adrenoceptor blockers have much less pronounced effects.
- Other drugs that can precipitate or aggravate hyperglycemia include:
Many drugs can cause or worsen pre-existing hyperglycemia – a problem recently recognized by the World Health Organization (WHO) and the American Diabetes Association (ADA) with the identification of drug-induced diabetes as a separate etiologic category (see Chapter 2). The diabetogenic properties of drugs are important for two main reasons. First, polypharmacy is an unfortunate but common necessity in managing patients with diabetes; clear understanding of the potential hyperglycemic effects of drugs is therefore helpful in anticipating and avoiding deterioration in glycemic control. Secondly, various drugs can induce diabetes in previously normoglycemic individuals; this state is usually reversible and not insulin-dependent, but can become permanent.
Drugs can raise blood glucose concentrations through two broad mechanisms: by reducing insulin biosynthesis or secretion, or by reducing tissue sensitivity to insulin (Figure 16.1). Some important examples are listed in Table 16.1. Of particular note are the glucocorticoids, which are used in many diseases, and certain commonly used antihypertensive drugs [1–7]. Hypertension commonly accompanies diabetes, and most patients require more than one antihypertensive agent to meet the increasingly stringent targets for blood pressure control (see the first part of Chapter 40).
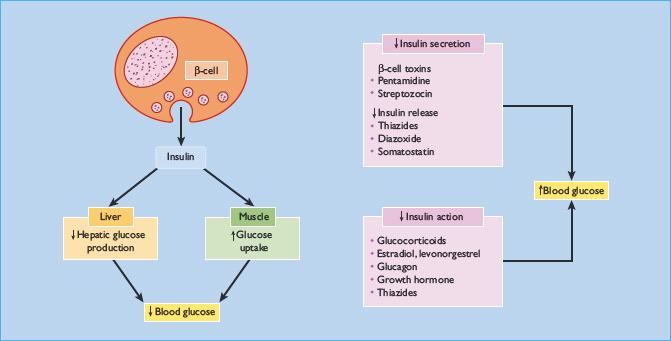
Table 16.1 Drugs that cause or exacerbate hyperglycemia.
| Potentially potent effects | Minor or no effects |
| Glucocorticoids | Oral contraceptives |
| Oral contraceptives | Progestogen-only pills |
| High-dose estrogen | Levonorgestrel in combination pills |
| Thiazide diuretics (especially high dosages)* | Loop diuretics |
| Non-selective β-adrenoceptor antagonists | Calcium-channel blockers |
| β2-adrenoceptor agonists | α1-adrenoceptor antagonists |
| Salbutamol | Growth hormone (physiologic doses) |
| Ritodrine | Somatostatin analogs† |
| Antipsychotics | Androgen deprivation therapy for prostate cancer |
| HIV protease inhibitors | Selective serotonin reuptake inhibitors |
| Indinavir, nelfi navir, ritonavir and others | Nicotinic acid |
| Lamivudine | |
| Isoniazid | |
| Others | |
| Pentamidine | |
| Gatifl oxacin | |
| Streptozocin (streptozotocin) | |
| Diazoxide | |
| Ciclosporin (cyclosporine) | |
| Tacrolimus | |
| Temsirolimus | |
| Interferon-α | |
| L-asparaginase |
HIV, human immunodefi ciency virus.
*“High” dosages of thiazides correspond to ≥5 mg/day bendroflumethiazide.
† Somatostatin analogs may induce hyperglycemia in type 2 but not type 1 diabetes.
This chapter describes the drugs that can induce or aggravate hyperglycemia, together with a strategy for managing patients with drug-induced diabetes.
Glucocorticoids
Glucocorticoids were named for their hyperglycemic effects [8] and have by far the most powerful adverse effect on glycemic control of all the commonly prescribed drugs. During the 1930s, it became apparent that diabetic symptoms improved following either adrenalectomy [9] or hypophysectomy [10], indicating that glucocorticoids have important influences on glucose homeostasis. This fact was recognized in clinical practice [11–15], soon after the landmark discovery in 1949 by Hench et al. [16] that glucocorticoids had potent anti-inflammatory effects. Since then, therapeutic use of these agents has escalated; recent data from the General Practice Research Database suggest that nearly 1% of the UK population may be using oral glucocorticoids at any time, increasing to 2.5% in subjects aged 70–79 years [17]. Inhaled corticosteroid use is even more widespread, with more than 6% of the UK population currently using an inhaled corticosteroid [18].
Glucocorticoids worsen hyperglycemia in patients with diabetes, but can also cause significant increases in blood glucose (and insulin) concentrations in previously normoglycemic individuals when given in high doses (i.e. equivalent to 30mg/day or more of prednisolone) [19]. Impaired glucose tolerance or diabetes mellitus have been reported in 14–28% of subjects receiving long-term glucocorticoids [20,21], and subjects who have an intrinsically low insulin response (e.g. in response to glucose loading) are particularly susceptible [22]. These subjects are thought to be at greater risk of developing type 2 diabetes (T2DM) in the future, compared to the general population [23]. Using data from the Health Improvement Network, Gulliford et al. [24] found that in a large primary care population, drawn from 114 family practices, orally administered glucocorticoids were associated with up to 2% of incident cases of diabetes mellitus.
Glucocorticoids reduce hepatic and peripheral tissue sensitivity to insulin through post-receptor mechanisms (Fig. 16.1). In adipocytes, dexamethasone inhibits the expression of the insulin-signaling intermediate protein, insulin receptor substrate 1 (IRS-1) [25], and this may contribute to insulin resistance (see Chapter 7). These effects may be partly offset by glucose-independent stimulation of insulin secretion [26].
All glucocorticoids cause dose-dependent insulin resistance at dosages greater than the equivalent of 7.5 mg/day prednisolone [21]. The duration of exposure to glucocorticoids does not appear to be important, and hyperglycemia is generally reversible on withdrawing the drug.
Most problems have been reported with oral glucocorticoids, but those administered topically can also induce severe hyperglycemia, especially if given at high dosage over large areas of damaged skin and under occlusive dressings [27]. This is more likely to occur in children because of their higher ratio of total body surface area to body weight [28]. Although inhaled corticosteroids do not cause significant hyperglycemia, there has been a single case report of deteriorating glycemic control in a patient with T2DM who was prescribed high-dose fluticasone propionate [29]. The hyperglycemic potency of glucocorticoids does not follow the hierarchy of their anti-inflammatory or immunosuppressive activities. For example, deflazacort, which has similar immunomodulating effects to other glucocorticoids, produces less hyperglycemia than prednisone or dexamethasone [30].
Other commonly encountered adverse effects of glucocorticoids are hypertension and sodium and water retention. Thiazide diuretics should not be used to treat these complications, as their own hyperglycemic action synergizes with that of glucocorticoids [31].
Corticotropin (adrenocorticotropic hormone [ACTH]) or tetracosactide (tetracosactrin), previously given as an alternative to glucocorticoid therapy (e.g. in exacerbations of multiple sclerosis), is no longer recommended for therapeutic use. It can cause hyperglycemia and hyperinsulinemia in rodents [32,33].
The metabolic adverse effects of glucocorticoids have stimulated the development of selective glucocorticoid receptor ligands with similar anti-inflammatory efficacy to glucocorticoids currently in use but with fewer adverse effects. A number of these compounds have been developed and shown in animal studies not to induce hyperglycemia [34]. These agents are likely to be tested clinically in humans in the near future.
Oral contraceptive pills and estrogen replacement therapy
Estrogens and some progestogens used in contraceptive agents are potentially diabetogenic. This is not surprising, as endogenous sex steroids have been shown to affect glucose homeostasis in women without diabetes: insulin sensitivity rises during the follicular phase of the menstrual cycle and falls during the luteal phase [35]. As with glucocorticoids, post-receptor insulin resistance appears to be responsible; in vivo studies have demonstrated a decrease in insulin sensitivity in women without diabetes taking certain contraceptive pills [36,37], and a number of implantable hormonal contraceptives have been linked to alterations in carbohydrate metabolism, including impaired glucose tolerance and increased insulin resistance [38,39]. The tendency to cause hyperglycemia was particularly marked with the early pills, which had a relatively high estrogen content; the overall risk of developing impaired glucose tolerance was 35% [40]. and even greater in women with a history of diabetes during pregnancy [41,42]. Impaired glucose tolerance during pregnancy remains a potent risk factor, even with the newer oral contraceptives; these women are three times more likely to develop diabetes with a progestogen-only pill than with a low-dose combined pill [43].
Most currently available combined oral contraceptives contain low estrogen dosage (25–50μg/day). In 1992, Rimm et al. [44] reported the metabolic effects of oral contraceptives in a large prospective survey of healthy volunteers, and found transient rises in serum insulin concentrations and impairment of glucose tolerance with all formulations. Consistent with this, metabolic studies have demonstrated fasting hyperinsulinemia and reduced insulin sensitivity [36,37,45]. Reassuringly, however, recent studies, including a massive prospective follow-up of almost 99 000 non-diabetic participants [46], found no appreciable increase in the incidence of diabetes among users of current oral contraceptives [46,47]. The authors of a recent Cochrane review [48] concluded that hormonal contraceptives have little clinical impact on carbohydrate metabolism. Low-dose combined oral contraceptives are safe in younger women with uncomplicated well-controlled diabetes [49].
In contrast to the older high-estrogen pills, the effect on glucose homeostasis of low-estrogen combination pills is determined mainly by the type and dosage of progestogen, with monophasic levonorgestrel combinations having the most deleterious effect. Oral progestogen-only formulations cause only minor hyperglycemia in healthy subjects, although diabetes may develop in women who had hyperglycemia during previous pregnancies [43]. The long-acting progestogens, such as depot medroxyprogesterone (Depo-Provera) and sustained-release low-dose levonorgestrel, cause a statistically but not clinically significant deterioration in glucose tolerance in healthy women [39,50]. There is little evidence, however, to suggest that this translates into clinical harm, and the WHO medical eligibility criteria for contraceptive use do not restrict the use of progestogen-only contraceptives in women with diabetes unless diabetes has been present for over 20 years or complications are present [51]. A primary care study in the UK found that progestogen-only methods of contraception, including the injectable contraceptive Depo-Provera and progestogen-only pills, are significantly more likely to be prescribed for women with diabetes than for those without diabetes [52]. There is no convincing evidence that the Mirena coil is associated with any change in glucose metabolism.
Hyperglycemia induced by hormonal contraceptives is usually reversible on withdrawal of the contraceptive pill. The current low-dosage contraceptive pills do not apparently predispose to the subsequent development of T2DM, although there is some evidence that this is a hazard in patients previously exposed to high-dosage pills [53].
Estrogen replacement therapy in women with diabetes
A number of observational studies of hormone replacement therapy (HRT) use in T2DM have demonstrated an association between HRT use and reduced cardiovascular risk [54,55]. This is in contrast to the general population, where HRT is not associated with a reduction in the incidence of cardiovascular disease in postmenopausal women [56]. HRT preparations differ with regard to type and dose of estrogen, type of combination progestogen and route of administration, and it is likely that these differences may be important in determining the overall balance of cardiovascular risk and benefit in specific patient subgroups such as those with diabetes. One placebo-controlled randomized study in 25 postmenopausal women with T2DM found that conjugated equine estrogen therapy (0.625mg/day) had beneficial effects on blood glucose and lipid profiles [57]. A further study in 28 postmenopausal women with T2DM using continuous oral 17β estradiol (1 mg) and norethisterone (0.5 mg) had similar findings [58]. In the Women’s Health Initiative study, involving over 160 000 postmenopausal women aged 50–79 years, those randomized to HRT had a lower incidence of self-reported diabetes than those randomized to placebo. This was true both for women taking conjugated equine estrogen alone and those taking a combination of conjugated equine estrogen plus medroxyprogesterone acetate [59,60]. The results of these studies, along with the findings of a meta-analysis quantifying the effect of HRT on components of the metabolic syndrome in postmenopausal women [61], provide some reassurance that postmenopausal estrogen replacement therapy can be offered to patients with diabetes.
Antihypertensive and cardiovascular agents
Thiazide diuretics
Hyperglycemia was recognized as an adverse effect of chlorothiazide (the first marketed thiazide diuretic) within a year of its introduction in 1957 [62]. Thiazides can induce adverse biochemical changes, including hyperglycemia, hypokalemia and dyslipidemia [63–68]. The overall frequency of impaired glucose tolerance is around 3–6 cases per 1000 patient-years when high dosages of thiazides (≥5 mg/day bendroflumethiazide (bendrofluazide) are used. With low dosages (≤2.5 mg/day bendroflumethiazide) and the correction of any fall in plasma potassium concentration, the problem is far less common [69–74].
Long-term studies show that impaired glucose tolerance usually develops slowly, with a lag period of 2 years after starting the thiazide [66,67]. The precise mechanism by which thiazide diuretics impair glucose tolerance has not been elucidated. It was previously thought to be secondary to both reduced glucose-stimulated insulin release and to insulin resistance (Figure 16.1), but some recent evidence suggests the mechanism involves reduced pancreatic insulin release alone and not reduced insulin sensitivity [75]. The severity of glucose intolerance is strongly correlated with the degree of hypokalemia [67]; the impairment of insulin secretion is secondary to potassium depletion, which appears to inhibit the cleavage of proinsulin to insulin and is reversible on restoring normokalemia [73,76,77]. The result is post-prandial hyperglycemia, with elevated proinsulin concentrations between meals that may downregulate insulin receptors in peripheral tissues. This phenomenon affects people without diabetes and those with T2DM, but not patients with type 1 diabetes (T1DM) receiving exogenous insulin replacement.
β-Adrenoceptor antagonists
β-Adrenoceptors modulate glucose homeostasis at several different points (Figure 16.2). Long-term studies suggest that β-adrenoceptor antagonists induce insulin resistance, possibly partly through weight gain [78]. This diabetogenic effect is amplified if high-dose thiazides are also given [79]. The authors of a recent meta-analysis of 94492 patients with hypertension concluded that treatment with β-adrenoceptor antagonists is associated with a 22% increased risk for new-onset diabetes [80]. The risk was greater in patients with higher baseline body mass indexes and higher baseline fasting glucose concentrations.
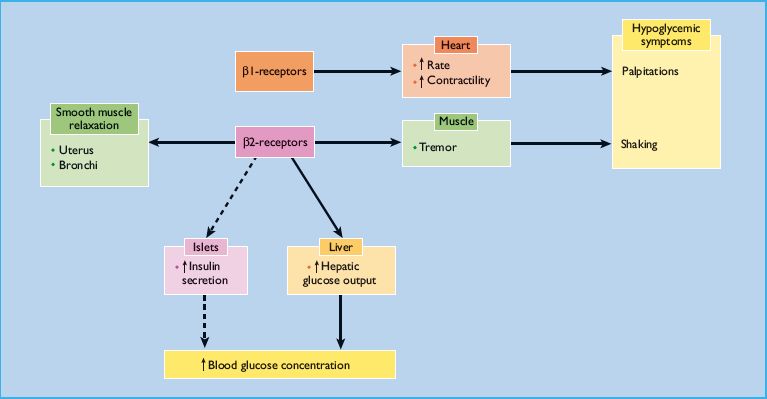
β-Adrenoceptor antagonists differ in their diabetogenic potential; in the meta-analysis, the risk for new-onset diabetes was 30% greater for atenolol and 34% greater with metoprolol than other agents [80]. In contrast, there was no increase in the risk for new-onset diabetes with propranolol compared with placebo. In fact, in the GEMINI trial, carvedilol stabilized glycated hemoglobin and improved insulin resistance in patients with diabetes and hypertension [81].
The possible importance of β-adrenoceptor antagonists in masking hypoglycemic symptoms is discussed in Chapter 26.
Calcium-channel blockers
In vitro and in vivo studies have demonstrated that calcium-channel blockers can impair glucose metabolism. Very few cases of clinically significant hyperglycemia have been reported, however, [82–88], and most were associated with excessive dosage of these drugs. Verapamil inhibits the second phase of glucose-stimulated insulin release by blocking the uptake of calcium into the cytosol of β-cells [89]; it also inhibits sulfonylurea and glucagon-induced insulin secretion [90]. Hyperglycemia and metabolic acidosis are well described in verapamil poisoning, and i n vivo animal studies suggest that hyperglycemia may be caused by a combination of impaired insulin release and insulin resistance, causing a fall in insulin-mediated glucose clearance, together with increased action of circulating catecholamines [91]. Serum glucose concentrations have been found to correlate directly with the severity of the calcium-channel blocker intoxication [92]. Nonetheless, calcium-channel blockers used appropriately can be regarded as safe in patients with diabetes [82,93–96].
Other drugs
β-Cell toxins
A few drugs act directly as β-cell toxins and can cause permanent diabetes, often insulin-dependent. A classic example is the nitrosourea, streptozocin (streptozotocin), which has long been used to induce experimental insulin-dependent diabetes in rodents; in humans, it is employed as chemotherapy for inoperable or meta-static insulinoma.
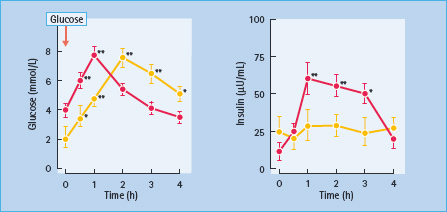
Pentamidine, used to treat Pneumocystis jirovecii infection in patients with AIDS [97], is closely related to another experimental diabetogenic drug, alloxan. It causes destruction of β-cells and may initially induce insulin release and transient hypoglycemia [98], followed subsequently by diabetes [99]. This effect is predominantly seen when pentamidine is given intravenously (Figure 16.3), but can also follow inhalation of an aerosol [100]. In one series of 128 patients with AIDS treated with pentamidine for Pneumocystis jirovecii infection, nearly 40% developed significant abnormalities of glucose homeostasis: hypoglycemia (5%), hypoglycemia followed by diabetes (15%) and diabetes alone (18%) [100]. Higher doses of pentamidine, higher plasma creatinine concentration and more severe anoxia were risk factors for the development of these glucose abnormalities.
Figure 16.3 Plasma glucose and insulin concentrations during an oral glucose tolerance test, in 20 healthy controls (red) and in four dysglycemic subjects treated with intravenous pentamidine (yellow). Data are mean ± SEM. * P < 0.05, ** P < 0.01 vs baseline. Reproduced from Assan et al. [100] with permission from the American Diabetes Association.
The rodenticide pyriminil (Vacor) has also caused irreversible diabetes in humans when ingested accidentally or with suicidal intent.
HIV protease inhibitors
The treatment of HIV infection has evolved dramatically during the last decade, with the wide introduction of protease inhibitors such as indinavir, nelfinavir, ritonavir and saquinavir. In general, these drugs are well tolerated, but worsening of pre-existing diabetes or the new onset of T2DM has been reported in 2–6% of patients receiving them [101–103]. Transient elevations in glucose concentrations are common in these patients, and there is a fivefold increase in the incidence of sustained glucose intolerance [101]. Because of similarities to the pathogenesis and clinical presentation of T2DM, it has been advised that treatment of protease inhibitor-associated hyperinsulinemia and hyperglycemia should follow the recommendations of the American Diabetes Association for the management of T2DM [104].
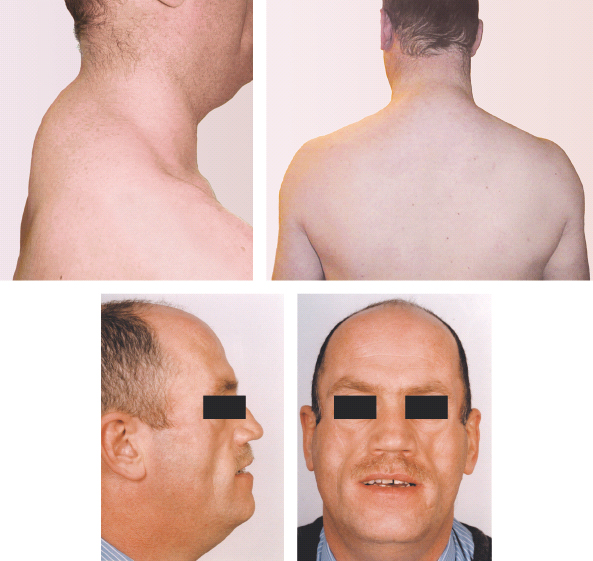
A striking syndrome of peripheral lipodystrophy, hyperlipidemia and insulin resistance has also been described in patients receiving HIV protease inhibitors, especially long term (Figure 16.4). This suggests that the underlying cause of hyperglycemia is insulin resistance [103]. The mechanism is not fully understood; Carr et al. [103] propose that protease inhibitors may bind to as yet uncharacterized target proteins that regulate lipid metabolism, leading to elevated circulating fatty acids that could interfere with insulin signaling or enter the fatty acid cycle and compete with glucose cycle intermediates. Recent studies indicate that these drugs have direct effects on adipocytes, including resistance to insulin’s actions in promoting glucose uptake and inhibiting lipolysis [105], as well as impairment of differentiation from pre-adipocytes to mature fat cells [106].
Figure 16.4 Protease inhibitor-induced lipodystrophy. (a,b) “Buffalo hump” caused by nuchal fat deposition. (c,d) Facial fat atrophy. Courtesy of Professor Munir Pirmohamed, University of Liverpool, UK.
β2-Adrenoceptor agonists
β2-Adrenoceptor agonists stimulate insulin secretion, but this effect is overwhelmed by increased hepatic glucose output, and the usual net effect is hyperglycemia (Figure 16.2). High dosage of β2-agonist drugs (e.g. salbutamol, ritodrine and terbutaline) is commonly used to treat asthma and premature labor. Hyperglycemia often results [107], and diabetic ketoacidosis has even been induced in previously non-diabetic pregnant women [108]. Continuous nebulization of β2-agonist drugs for status asthmaticus can also result in hyperglycemia [109]. The effects of β2-agonists are most pronounced in patients with T1DM. When dexamethasone is administered together with a β2-agonist, as in the treatment of preterm labor, the resulting hyperglycemia can be severe, even in previously normoglycemic patients. In view of this, current guidance from the National Institute for Health and Clinical Excellence (NICE) recommends that when tocolysis is indicated in women with diabetes, an alternative to β2-adrenoceptor agonists should be used to avoid hyperglycemia and ketoacidosis [110].
Epinephrine (adrenaline) [111], dopamine [112] and theophylline can also cause hyperglycemia through similar mechanisms.
Diazoxide
Diazoxide is a non-diuretic benzothiadiazine derivative with potent vasodilator properties, which was previously used to treat hypertensive crises; it is now rarely used for this indication, but is sometimes helpful in cases of inoperable insulinoma and to treat profound hypoglycemia following sulfonylurea overdose. The dose used for treatment of patients with insulinoma ranges between 100 and 600 mg/day, often in divided doses [113]. It acts on the β-cell membrane to open the ATP-dependent potassium channel, thus hyperpolarizing the membrane and inhibiting insulin secretion (see Chapter 6). Diazoxide can cause hyperglycemia after only two or three doses.
Somatostatin analogs
Somatostatin suppresses insulin secretion but also inhibits the release of the counter-regulatory hormones, growth hormone and glucagon; the net effect in subjects without diabetes is usually to maintain euglycemia. Octreotide, a somatostatin analog used to treat neuroendocrine tumors, has different metabolic effects in T1DM and T2DM [114]. In patients with T1DM treated with exogenous insulin, suppression of glucagon and growth hormone decreases hepatic glucose production and can lower blood glucose and/or insulin requirements. In patients with T2DM, inhibition of endogenous insulin can predominate, leading to hyperglycemia. There is some evidence that treatment with octreotide, lanreotide and the long-acting preparations of both drugs can impair glucose tolerance and occasionally cause diabetes, notably in people with acromegaly whose glucose homeostasis is already impaired (see Chapter 17) [115–117]. The effect of long-acting somatostatin analogs on glucose metabolism in patients with acromegaly is complex; they reduce the insulin resistance caused by elevated growth hormone concentrations, but also suppress insulin secretion from islet β-cells. The net balance between the two effects determines whether long-acting somatostatin analogs improve or worsen glucose metabolism [118]. These effects are inconsistent and unpredictable, with worsening glucose metabolism occasionally seen in the presence of improving growth hormone concentrations [119].
Utilizing its suppressive effect on insulin release, octreotide has been used effectively to manage refractory hypoglycemia caused by acute sulfonylurea poisoning or quinine treatment [120–123].
Recombinant human growth hormone
Biosynthetic growth hormone (GH), now widely used to treat growth failure, is an insulin antagonist that can cause reversible mild glucose intolerance [124]. A recent large study of over 23 000 children treated with human recombinant human growth hormone (hGH) showed a sixfold higher frequency of T2DM than expected [125], although the incidence of T1DM was not increased. These data from a young cohort suggest that GH probably accelerates the development of the disease in individuals who may have a genetic predisposition to T2DM.
Increasingly, hGH has become accepted as long-term therapy in adults with GH deficiency (GHD) (see Chapter 17); as yet, no significant adverse effects on glycemic control have been reported. Fasting plasma glucose and glycated hemoglobin usually increase, but remain within the reference range, in subjects without diabetes during prolonged hGH replacement; in subjects with preexisting impaired glucose tolerance, they tend not to rise further above pretreatment baseline values [126]. This probably reflects the complex interaction between hGH, insulin-like growth factor I (IGF-I) and body composition. Many people with adult GHD have abnormal body composition with central adiposity and decreased lean body mass. In the short term, rhGH acts as an insulin antagonist but in the longer term leads to increases in IGF-I and decreased fat mass, which tend to improve insulin sensitivity (see Chapter 17). It is recommended, however, that overdosing be avoided and glycemic control be monitored [127].
New-onset diabetes mellitus has been described in patients with the HIV wasting syndrome (AIDS cachexia) treated with hGH in supraphysiological doses [128].
Antirejection drugs
Post-transplantation diabetes mellitus (PTDM) has been reported in non-diabetic adult transplant recipients taking ciclosporin (cyclosporine) [129,130]. Animal studies have implicated reversible β-cell damage [131], causing reduced insulin secretion [132–134]. In addition, pancreas allograft biopsies from transplant patients receiving ciclosporin show histologic changes compatible with islet cell damage, including cytoplasmic swelling, vacusolization and apoptosis [135]. Recent studies suggest that the risk of PTDM increases progressively with time after transplantation. Newer preparations of ciclosporin which are better absorbed from the gastrointestinal tract achieve higher blood concentrations and thus higher cumulative exposure, and increase the incidence of diabetes [136].
PTDM is also commonly observed in up to 28% of adults receiving tacrolimus (FK506), a newer and more potent immunosuppressive agent [137–139]. More recent trials, however, suggest the risk of developing PTDM can be reduced by the use of low-dose tacrolimus (0.15–0.2 mg/kg/day) [140]. Altered insulin and glucagon responses to arginine in patients treated with these drugs suggest a defect in the β-cell–α-cell axis within the islet (see Chapter 6) [141]. The diabetogenic effects of both ciclosporin and tacrolimus are largely reversible with appropriate reductions in the drug dosage.
Recently developed immunosuppression protocols aiming to minimize the use of steroids and nephrotoxic immunosuppressants have resulted in the extensive use of potent non-nephrotoxic immunosuppressants, such as mycophenolate mofetil and sirolimus. The incidence of PTDM associated with these agents is uncertain. Study results with sirolimus have been inconsistent, with some suggesting an increased risk of developing PTDM [142,143] and others not [144].
α-Interferon therapy has been associated with the development of both T1DM and T2DM, sometimes with ketoacidosis; autoimmune mechanisms have been implicated [145].
Drugs used in psychiatric disorders
Antipsychotic agents
Hyperglycemia occurs occasionally with conventional antipsychotic drugs, but the use of the newer atypical antipsychotics, especially clozapine and olanzapine, have been widely reported to be associated with the development of de novo diabetes mellitus and exacerbation of pre-existing diabetes [146–148]. A causative relationship between antipsychotic and diabetes has not been established beyond doubt because many patients receiving these drugs who develop diabetes have traditional risk factors for diabetes. Indeed, the rates of diabetes in people with severe mental illness were reported to be higher in the pre-antipsychotic era (see Chapter 55). Possible underlying mechanisms linking antipsychotics and the development of diabetes include hepatic dysregulation caused by antagonism of hepatic serotonergic mechanisms [149]. Weight gain, associated with fasting hyperglycemia and hyperinsulinemia, point to insulin resistance as the underlying mechanism although some in vitro studies suggest that the antipsychotics may have a direct effect on insulin secretion. In a few cases, blood glucose concentrations may return to normal once the drug is discontinued. Despite a wealth of evidence from a number of sources (anecdotal case reports, drug safety studies, pharmacoepidemiologic studies, prospective studies) linking glucose intolerance to the use of atypical antipsychotics, one estimate of the attributable risk of diabetes associated with atypical antipsychotics ranged from 0.05% for risperidone to 2.03% for clozapine, suggesting that the absolute excess risk associated with atypical antipsychotics is probably low [150]. Consequently, most individuals receiving antipsychotics will not develop diabetes and for those who do, the cause is unlikely to be related to their treatment. When prescribing atypical antipsychotic medication, however, baseline screening and follow-up monitoring is recommended.
Antidepressants
Depression is an important problem among people with diabetes (see Chapter 55), and various antidepressant drugs can affect plasma glucose and insulin concentrations [151,152]. The tricyclic antidepressant, nortriptyline, worsens glycemic control [153], and has been shown to reduce insulin concentrations in animal studies [152]. There is also a single case report in the literature of another tricyclic agent, clomipramine, causing significant symptomatic hyperglycemia which resolved when the drug was discontinued and recurred when the patient was rechallenged with the drug [154]. The selective serotonin reuptake inhibitors (SSRIs), fluoxetine and fluvoxamine, cause hyperglycemia in mice, apparently because increased serotonergic neurotransmission enhances catecholamine release from the adrenal medulla while suppressing insulin release [151]. This does not, however, appear to be a significant problem in humans; indeed, various antidepressant drugs, including SSRIs, have been shown to improve glycemic control in patients with diabetes by decreasing appetite while relieving depression and helping to improve compliance with antidiabetic treatment [155].
Other drugs
- Asparaginase (crisantaspase), an anticancer drug used to treat acute lymphoblastic leukemia, causes predictable impairment of glucose tolerance which is secondary to insulin resistance. In one trial in children, 10% of cases developed hyperglycemia, and all showed glycosuria [156].
- Oxymetholone and danazol, certain synthetic steroid derivatives with androgenic properties, impair glucose tolerance, probably by inducing insulin resistance at a post-receptor site [157]; stimulation of glucagon secretion may also contribute [158].
- Nicotinic acid, used to treat dyslipidemia, is reported to cause hyperglycemia which is occasionally severe [118,159]. although no such deleterious effect was observed in a recent trial in people with diabetes [160]. Its analog, acipimox, does not have adverse effects on glycemic control in people with diabetes [161].
- Phenytoin can cause hyperosmolar hyperglycemic syndrome or overt diabetes by interfering with calcium ion entry into β-cells, thus inhibiting insulin secretion [162].
- Gatifloxacin is a broad-spectrum 8-methoxyfluoroquinolone antibacterial agent previously used to treat a variety of infections. There have been several postmarketing reports of dysglycemia, both hypoglycemia and hyperglycemia, associated with the use of gatifloxacin [163,164]. The incidence of gatifloxacin-induced hyperglycemia is estimated at around 1%, and reported cases have involved both new-onset diabetes and worsening of glycemic control in patients with existing diabetes. The exact underlying mechanism is unknown, but data from animal studies point towards possible inhibition of insulin secretion [164] or increased secretion of epinephrine [165].
Transient hyperglycemia has been described following treatment or overdose with a number of commonly prescribed drugs such as non-steroidal anti-inflammatory drugs [166] and isoniazid. There are also anecdotal reports of drug-induced hyperglycemia associated with nalidixic acid [167], carbamazepine [168], encainide [169], benzodiazepines [170] and mianserin [171].
Treatment of drug-induced hyperglycemia
Clinically relevant hyperglycemia occurs most commonly with high doses of glucocorticoids. If hyperglycemia occurs during thiazide treatment, the need for the drug should be reassessed. If a diuretic is required, then a small dose of furosemide or bumetanide may be substituted. If an antihypertensive agent is needed, it may be possible to reduce the dosage of bendroflumethiazide (e.g. to 2.5 mg/day), or to replace it with another class of drug.
Steroid-induced diabetes
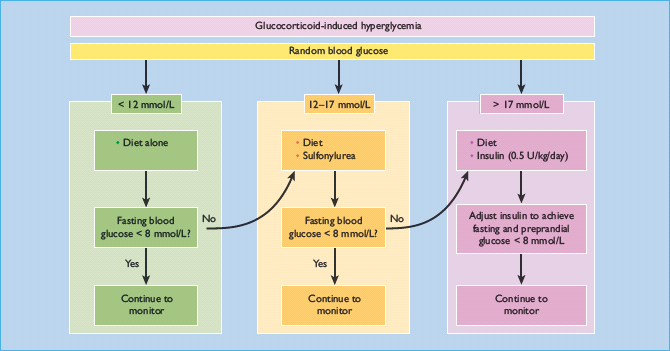
It may not be possible to withdraw glucocorticoid therapy, although “steroid-sparing” immunosuppressive drugs such as azathioprine can sometimes be introduced for certain indications. Other measures are often needed to control hyperglycemia. Management depends primarily upon the severity of hyperglycemia (Figure 16.5). Random blood glucose measurements provide only an approximate guide, and therapy should be adjusted by frequent blood glucose monitoring; simple relief of symptoms alone is inadequate. A target fasting blood glucose concentration of <8 mmol/L may be suitable in the short term, and the usual criteria for good control should be applied if long-term glucocorticoid therapy is undertaken (see Chapter 20).
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


