INTRODUCTION
SUMMARY
When procoagulants are produced or introduced into the blood and overcome the anticoagulant mechanisms of coagulation, intravascular thrombin is generated systemically, which can lead to disseminated intravascular coagulation (DIC). The clinical manifestations of intravascular coagulation include (1) multiorgan dysfunction caused by microthrombi; (2) bleeding caused by consumption of platelets, fibrinogen, and other coagulation factors; and (3) secondary fibrinolysis. Exposure of blood to tissue factor is the most common trigger. This event can occur when mononuclear cells and endothelial cells are induced to generate and express tissue factor during the systemic inflammatory response syndrome (e.g., Gram-negative and Gram-positive infections, fungemia, burns, severe trauma), or when contact is established between blood and tissue factor constitutively present on membranes of cells foreign to blood (e.g., malignant, placental, brain, adventitial cells, or traumatized tissues). Laboratory features include thrombocytopenia, reduced levels of fibrinogen and other coagulation factors (leading to prolonged partial thromboplastin, prothrombin, and thrombin times), and elevated levels of D-dimer and fibrin(ogen) degradation products. Several underlying disorders affect these hemostatic parameters and can lead to a false-positive diagnosis of DIC (e.g., liver disease–related coagulation abnormalities and thrombocytopenia) or to a false-negative diagnosis (e.g., pregnancy-related high fibrinogen levels). Reexamining these variables every 6 to 8 hours may permit a specific diagnosis. Early detection, vigorous treatment of the underlying disorder, and support of vital functions are essential for survival of affected patients. Blood component therapy is effective in patients who bleed excessively, whereas heparin administration is indicated in a limited number of circumstances. Intravascular coagulation and the underlying disorders causing it contribute to a high rate of mortality. The severity of the organ dysfunction and extent of hemostatic failure, as well as increasing patient age, have been associated with a grave prognosis.
Acronyms and Abbreviations
APACHE, acute physiology and chronic health evaluation; APC, activated protein C; APL, acute promyelocytic leukemia; aPTT, activated partial thromboplastin time; ARDS, adult respiratory distress syndrome; AT, antithrombin; DIC, disseminated intravascular coagulation; EPCR, endothelial protein C receptor; FDP, fibrinogen degradation product; HELLP, hemolysis, elevated liver enzymes, low platelet count; IL, interleukin; LCAD, long-chain acyl-coenzyme A dehydrogenase; LPS, lipopolysaccharide; PAI, plasminogen-activator inhibitor; PAR, protease-activated receptor; TAFI, thrombin-activatable fibrinolysis inhibitor; TAT, thrombin–antithrombin; TF, tissue factor; TFPI, tissue factor pathway inhibitor; TNF, tumor necrosis factor; t-PA, tissue-type plasminogen activator.
DEFINITION AND HISTORY
Disseminated intravascular coagulation (DIC) is a clinicopathologic syndrome in which widespread intravascular coagulation occurs as a result of exposure or production of procoagulants insufficiently balanced by natural anticoagulant mechanisms and endogenous fibrinolysis. Perturbation of the endothelium in the microcirculation along with stimulated inflammatory cells and release of inflammatory mediators play a key role in this mechanism. DIC may cause tissue ischemia from occlusive microthrombi, and bleeding from the consumption of platelets and coagulation factors and, in some cases, an excessive fibrinolytic response. DIC complicates a variety of disorders, and the complexity of its pathophysiology has made it the subject of a voluminous literature.1,2,3,4,5,6,7
In 1834, Dupuy reported that injection of brain material into animals caused widespread clots in blood vessels, thus providing the first description of DIC.8 In 1865, Trousseau described the tendency to thrombosis, sometimes disseminated, in cachectic patients with malignancies.9 In 1873, Naunyn showed that disseminated thrombosis could be evoked by intravenous injection of dissolved red cells, and Wooldridge demonstrated that the procoagulant involved was a substance contained in the stroma of the red cells.10,11,12
In 1955, Ratnoff and associates described the hemostatic abnormalities, which we would currently classify as DIC, that occur in women with fetal death or amniotic fluid embolism.13 The mechanism by which DIC can lead to bleeding was clarified only in 1961, when Lasch and coworkers introduced the concept of consumption coagulopathy, and McKay established that DIC is a pathogenetic feature of a variety of diseases.1,14 Sizable series of cases were first described in the late 1960s, following the introduction of defined laboratory criteria for DIC.15 Yet despite the vast experience that has been accumulated, DIC still constitutes a major clinicopathologic and therapeutic challenge.
PATHOLOGY
Diffuse multiorgan bleeding, hemorrhagic necrosis, microthrombi in small blood vessels, and thrombi in medium and large blood vessels are common findings at autopsy, although patients who had unequivocal clinical and laboratory signs of DIC may not have had confirming postmortem findings.16,17 Conversely, some patients in whom clinical and laboratory signs were not consistent with DIC had typical autopsy findings.18,19 This occasional lack of correlation among clinical, laboratory, and pathologic findings is partly a result of extensive postmortem changes in the blood, for example, excessive fibrinolysis, but remains unexplained in most instances.17 Organs most frequently involved by diffuse microthrombi are the lungs and kidneys, followed by the brain, heart, liver, spleen, adrenal glands, pancreas, and gut. Specific immunohistologic techniques and ultrastructural analysis have revealed that most thrombi consist of fibrin monomers or polymers in combination with platelets. In addition, involvement of activated mononuclear cells and other signs of inflammatory activation are frequently present.20 In cases of long-lasting DIC, organization and endothelialization of the microthrombi are often observed. Acute tubular necrosis is more frequent than renal cortical necrosis.16
A significant proportion of patients with chronic DIC have nonbacterial thrombotic endocarditis involving mainly the mitral and aortic valves.19 Moreover, in a retrospective pathologic study, approximately 50 percent of patients with nonbacterial thrombotic endocarditis had DIC.18 These heart lesions can be a source of arterial embolization, leading to infarction of the brain, kidneys, and myocardium.
PATHOGENESIS
Various triggers cause an hemostatic imbalance that gives rise to a procoagulant state (Fig. 129–1). The most important mediators responsible for this imbalance are cytokines.21 There is an extensive crosstalk between coagulation and inflammatory systems, whereby inflammation leads to activation of coagulation, and coagulation stimulates inflammatory activity.22 These interactions are highlighted in sepsis-induced systemic activation of coagulation and inflammation that lead to specific organ dysfunctions.23 The endothelium of the capillary bed is the most important interface in which the interaction between inflammation and coagulation takes place. Endothelial cells may be a source of tissue factor and can thereby be involved in the initiation of coagulation activation. All physiologic anticoagulant systems and various adhesion molecules that may modulate both inflammation and coagulation are connected to the endothelium. In sepsis, endothelial glycosaminoglycans present in the glycocalyx are downregulated by proinflammatory cytokines, thereby impairing the functions of antithrombin (AT), tissue factor pathway inhibitor (TFPI), leukocyte adhesion, and leukocyte transmigration. Because the glycocalyx also plays a role in other endothelial functions, including maintenance of the vascular barrier function, nitric oxide–mediated vasodilation, and antioxidant activity, all these processes can be impaired in DIC (see “Role of Oxidative Stress and Vasoactive Molecules” below).24,25 Moreover, specific disruption of the glycocalyx results in thrombin generation and platelet adhesion within a few minutes.26,27
Figure 129–1.
Schematic presentation of pathogenetic pathways involved in the activation of coagulation in disseminated intravascular coagulation (DIC). In DIC, both perturbed endothelial cells and activated mononuclear cells may produce proinflammatory cytokines that mediate coagulation activation. Activation of coagulation is initiated by tissue factor expression on activated mononuclear cells and endothelial cells. In addition, downregulation of physiologic anticoagulant mechanisms and inhibition of fibrinolysis by endothelial cells further promote intravascular fibrin deposition. PAI-1, plasminogen-activator inhibitor type 1.
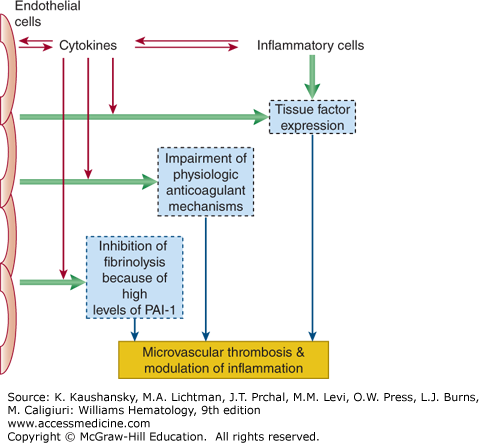
Endothelial perturbation constitutes a sine qua non for most patients with DIC. Following injury or infection, the integrity of the endothelium is compromised, mononuclear cells are activated by cytokine and hormonal signals, additional cytokines and surface receptors are upregulated, procoagulant proteins and platelets are activated, the endothelium changes from an anticoagulant to procoagulant surface, and fibrinolysis is impeded. This sequence of events is typical for the systemic inflammatory response syndrome and can lead to microvascular thrombosis with ensuing multiorgan dysfunction and eventually to multiorgan failure.
Tissue factor (TF) plays a central role in the initiation of inflammation-induced coagulation in DIC.28 Blocking TF activity completely inhibits inflammation-induced thrombin generation in experimental models of endotoxemia or bacteremia.29,30 Most cells constitutively expressing TF are found in tissues not in direct contact with blood, such as the adventitial layer of larger blood vessels. TF becomes exposed to blood upon disruption of the vascular integrity, or when cells present in the circulation, such as monocytes, are triggered to express TF. The in vivo expression of TF is dependent on interleukin (IL)-6 generation; inhibition of IL-6, unlike inhibition of other proinflammatory cytokines, completely abrogates TF-dependent thrombin generation in experimental endotoxemia.21,31 In severe sepsis, monocytes, stimulated by proinflammatory cytokines, express TF, which leads to systemic activation of coagulation.32 Even in experimental low-dose endotoxemia in healthy subjects, a 125-fold increase in TF mRNA levels in blood monocytes can be detected.33 A potential alternative source of TF may be endothelial cells, polymorphonuclear cells, and other cell types. It is hypothesized that TF from these sources is shuttled between cells through microparticles derived from activated mononuclear cells.34 However, it is unlikely that cells other than monocytes synthesize TF in substantial quantities.32,35 Tumor necrosis factor (TNF)-α and IL-1, also generated during inflammation, impair the physiologic anticoagulant pathways.31,36,37
The TF–factor VIIa complex catalyzes the conversion of factor X to Xa, and factor Xa, in turn, forms the prothrombinase complex with factor Va, prothrombin (factor II), and calcium ions, thereby generating thrombin, and converting fibrinogen into fibrin. The TF–factor VIIa complex can also activate factor IX, and factor IXa forms the tenase complex with activated factor VIII and calcium ions, generating additional factor Xa, thereby forming an essential amplification loop of thrombin generation. The assembly of the prothrombinase and tenase complexes are markedly facilitated if a suitable phospholipid surface is available, such as the membrane of activated platelets. In the setting of inflammation-induced activation of coagulation, platelets can be activated directly by endotoxin or by proinflammatory mediators, such as the membrane of platelet-activating factor. Thrombin itself is one of the strongest platelet activators (Chap. 115).
Activation of platelets may also accelerate fibrin formation by another mechanism. The expression of TF on monocytes is markedly stimulated by the presence of platelets and granulocytes in a P-selectin–dependent reaction.38 This effect may be the result of nuclear factor kappa B (NF-κB) activation induced by binding of activated platelets to neutrophils and mononuclear cells.39 This cellular interaction also markedly enhances the production of IL-1β, IL-8, monocyte chemotactic protein (MCP)-1, and TNF-α.40
Thrombin generated by the TF pathway amplifies both clotting and inflammation through the following activities: (1) it activates platelets, giving rise to platelet aggregation and augmenting platelet functions in coagulation; (2) it activates factors VIII, V, and XI, yielding further thrombin generation; (3) it activates proinflammatory factors via protease-activated receptors (PARs); (4) it activates factor XIII to factor XIIIa, which crosslinks fibrin clots; (5) it activates thrombin-activatable fibrinolysis inhibitor (TAFI), making clots resistant to fibrinolysis; and (6) it increases expression of adhesion molecules, such as L-selectin, thereby promoting the inflammatory effects of leukocytes.41
Paradoxically, at low concentrations, thrombin exhibits both antiinflammatory and anticoagulant effects because it binds to thrombomodulin and activates protein C to the activated form, which, in turn, downregulates inflammation and serves as an “off switch” for further thrombin generation (Chap. 116).
Coagulation proteases and protease inhibitors not only interact with coagulation proteins, but also with specific cell receptors to induce signaling pathways. In particular, protease interactions that affect inflammatory processes may be important in critically ill patients. Coagulation of whole blood in vitro results in a detectable expression of IL-1β mRNA in blood cells,42 and thrombin markedly enhances endotoxin-induced IL-1 activity in culture supernatants of guinea pig macrophages.43 Similarly, clotted blood produces IL-8 in vitro.44
Factor Xa, thrombin, and fibrin can also activate endothelial cells, eliciting the synthesis of IL-6 and IL-8.45,46 Coagulation proteases such as thrombin, factor Xa, and factor VIIa–TF complex induce inflammatory upregulation via leukocyte, endothelial cell, and platelet PAR-1, PAR-2, PAR-3, and PAR-4, which are located on leukocytes, endothelial cells, and platelets.47 PARs have an extracellular domain, seven transmembrane domains, and an intracellular domain that is coupled to specific G-proteins that transmit signaling. PAR-1, PAR-3, and PAR-4 are activated by thrombin through cleavage of a specific aminoterminus bond creating a tethered ligand that activates the receptor. PAR-2 can be cleaved by factor Xa–TF–factor VIIa complex and by other proteases.48 Activated PARs then lead through mitogen-activated protein kinase and NF-κB signaling pathways to cell motility, shape change, proliferation, endogenous secretagogue release, and apoptosis. The activated protein C (APC)–endothelial protein C receptor complex (see “Role of Natural Anticoagulant Pathways” below) appears to be the “off switch” for PAR activation by the proteases. These counterbalances determine the magnitude of coagulation and inflammatory upregulation by PARs. For example, factor VIIa–TF binding to PAR-2 in the lungs is proinflammatory and appears to play a role in acute respiratory distress syndrome (ARDS), raising the possibility that TFPI might be therapeutic in this circumstance.49 This finding is consistent with data from animal studies demonstrating that TFPI can protect baboons from a LD100 of Escherichia coli, likely by impeding factor VIIa–TF activation of PAR-2 and thereby attenuating release of IL-6 and other proinflammatory agents.
Fibrinogen and fibrin directly influence the production of proinflammatory cytokines and chemokines (including TNF-α, IL-1β, and MCP-1) by mononuclear cells and endothelial cells.50 Fibrinogen-deficient mice display inhibition of macrophage adhesion and less thrombin-mediated cytokine production in vivo. The effects of fibrinogen on mononuclear cells seem to be mediated by toll-like receptor-4, which is also the receptor of endotoxin.
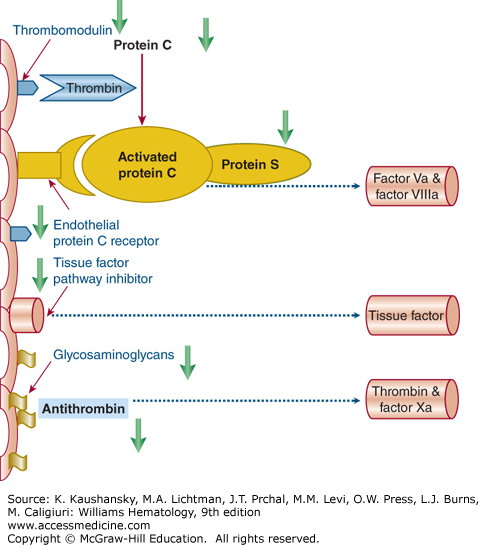
Procoagulant activity is regulated by three important anticoagulant pathways: AT, the protein C system, and TFPI. In DIC, the function of all three pathways can be impaired (Fig. 129–2).51
Figure 129–2.
Schematic of the three important physiologic anticoagulant mechanisms and their point of impact in the coagulation system. In sepsis, these mechanisms are impaired by various mechanisms (green arrows). The protein C system is dysfunctional as a result of low levels of zymogen protein C, downregulation of thrombomodulin and the endothelial protein C receptor, and low levels of free protein S because of acute phase-induced high levels of its binding protein (i.e., C4b-binding protein). There is a relative insufficiency of the endothelial cell-associated tissue factor pathway inhibitor. The antithrombin system is defective because of low levels of antithrombin and impaired glycosaminoglycan expression on perturbed endothelial cells.
The serine protease inhibitor AT is the main inhibitor of thrombin and factor Xa. Without heparin, AT neutralizes coagulation enzymes in a slow, progressive manner.52 Heparin induces conformational changes in AT that result in at least a 1000-fold enhancement of AT activity. Thus, the clinical efficacy of heparin is attributed to its interaction with AT. Endogenous glycosaminoglycans, such as heparan sulfate, also promote on the vessel wall AT-mediated inhibition of thrombin and other coagulation enzymes. During severe inflammatory responses, AT levels are markedly decreased because of impaired synthesis, degradation by elastase from activated neutrophils, and consumption as a consequence of ongoing thrombin generation.53 Proinflammatory cytokines also cause reduced synthesis of glycosaminoglycans on the endothelial surface, thereby reducing AT function.54
APC appears to play a central role in the pathogenesis of sepsis and associated organ dysfunction.55 There is ample evidence that decreased function of the protein C pathway contributes to the derangement of coagulation in sepsis.49,56 The circulating zymogen protein C is activated by thrombin when it is bound to thrombomodulin at the endothelial cell surface.57 APC acts with its cofactor protein S and degrades the essential cofactors Va and VIIIa, and hence, is an effective anticoagulant. The endothelial protein C receptor (EPCR) accelerates the activation of protein C several-fold, and also serves as a receptor for APC, thereby augmenting APC’s anticoagulant and antiinflammatory activities.58
In patients with severe inflammation, the protein C pathway malfunctions at virtually all levels. Plasma levels of the zymogen protein C are decreased because of impaired synthesis, consumption, and degradation by proteolytic enzymes, such as neutrophil elastase.59,60,61 Furthermore, a significant downregulation of thrombomodulin, caused by proinflammatory cytokines such as TNF-α and IL-1, results in diminished protein C activation.62,63 Low levels of free protein S may further compromise the function of the protein C system. In plasma, 60 percent of protein S is complexed with a complement regulatory protein, C4b-binding protein (C4bBP), and exhibits no activity. The remaining protein S in plasma is free and functional. It was suggested that increased plasma levels of C4bBP caused by the acute phase reaction in inflammatory diseases results in a relative protein S deficiency, which further contributes to a procoagulant state during sepsis. Indeed, infusion of C4bBP in combination with a sublethal dose of E. coli into baboons resulted in a lethal response with severe organ damage because of DIC.64
In sepsis, the EPCR is downregulated, which further impairs the function of the protein C pathway.65 Sepsis can also cause resistance toward APC because of a substantial increase in factor VIII levels.66
A third inhibitory mechanism of thrombin generation involves TFPI, the main inhibitor of the TF–factor VIIa complex and factor Xa. The role of TFPI in the regulation of inflammation-induced coagulation activation is not completely clear. Administration of recombinant TFPI blocks inflammation-induced thrombin generation in humans, and pharmacologic doses of TFPI prevent mortality during systemic infection and inflammation in experimental animals suggesting that TFPI can modulate TF-mediated coagulation.67,68
AT possesses antiinflammatory properties, many of which are mediated by its actions in the coagulation cascade.69 By inhibiting thrombin, AT blunts activation of many inflammatory mediators released by platelets and endothelial cells that recruit and activate leukocytes.70 At high concentrations, AT also possesses potent antiinflammatory properties that are independent of its anticoagulant activity.70 Another effect of AT is the induction of prostacyclin release from endothelial cells.71,72,73 Prostacyclin inhibits platelet activation and aggregation, blocks neutrophil tethering to blood vessels, and decreases endothelial cell production of various cytokines and chemokines.74
AT also interacts directly with leukocytes and lymphocytes. It binds to receptors, such as syndecan-4, on the cell surfaces of neutrophils, monocytes, and lymphocytes, thereby blocking the adhesion of these cells to endothelial cells, their activation and migration. This effect, in turn, ameliorates the severity of capillary leakage and subsequent organ damage.
The protein C system also has an important function in modulating inflammation.75,76 Blocking the protein C pathway in septic baboons exacerbates the inflammatory response, and in contrast, administration of APC ameliorates the inflammatory activation upon the intravenous infusion of E. coli.77 Support for the notion that APC has antiinflammatory properties comes from in vitro observations, demonstrating an APC binding site on monocytes, that may mediate downstream inflammatory processes,78,79 and from experiments showing that APC can block NF-κB nuclear translocation, which is a prerequisite for increased proinflammatory cytokine levels and adhesion molecules.80 These in vitro findings are supported by in vivo studies in mice with targeted disruption of the protein C gene. In these mice with genetic deficiencies of protein C, endotoxemia was associated with a more marked increase in proinflammatory cytokines and other inflammatory responses as compared with wild-type mice.81,82
It is likely that the antiinflammatory effects of APC are mediated by the EPCR.75 Binding of APC to EPCR influences gene expression profiles of cells by inhibiting NF-κB nuclear translocation.79,80 The EPCR-APC complex itself can translocate from the plasma membrane into the cell nucleus, which may be another mechanism of modulating gene expression, although the relative contribution of this nuclear translocation and cell surface signaling is uncertain.56 Like APC, EPCR itself may have antiinflammatory properties. Blocking the EPCR with a specific monoclonal antibody aggravates both the coagulation and the inflammatory response to E. coli infusion.65
Apart from the effect on cytokine levels, APC causes diminished leucocyte chemotaxis and adhesion to the activated endothelium.83,84,85 A localized antiinflammatory effect of APC has been demonstrated in the lung.86 One mechanism for this effect may be inhibition of the expression of platelet-derived growth factor in the lung.87 Also, APC protects against the disruption of endothelial cell barrier in sepsis.88,89,90 APC also inhibits endothelial cell apoptosis by a mechanism that seems to be mediated by binding of APC to EPCR and requires PAR-1.91,92 Signaling through this pathway can affect Bcl-2 homologue protein, which can inhibit apoptosis, and further suppresses p53, that is a proapoptotic transcription factor.93,94
In experimental models of DIC, fibrinolysis is initially activated but subsequently inhibited, because of an increased release of plasminogen activator inhibitor-I (PAI-1) by endothelial cells.95 These effects are mediated by TNF-α and IL-1.96,97 In a study of 69 DIC patients (31 with multiorgan failure), higher levels of tissue-type plasminogen activator (t-PA) antigen and PAI-1 with depressed levels of α2-antiplasmin were observed in patients with DIC and multiorgan failure compared to DIC patients without multiorgan failure.98 This finding supports the conclusion that fibrinolysis is an important mechanism in preventing multiorgan failure.
Experiments in mice with targeted disruptions of genes encoding components of the plasminogen–plasmin system confirm that fibrinolysis plays a major role in inflammation. Mice with a deficiency of plasminogen activators have more extensive fibrin deposition in organs when challenged with endotoxin, whereas PAI-1 knockout mice, in contrast to wild-type controls, have no microvascular thrombosis upon endotoxin administration.99
TAFI, like PAI-1, may play a role in impeding fibrinolysis and in augmenting formation of microvascular thrombi. Studies in a DIC cohort demonstrated very low levels of TAFI proportionate to thrombin generation in such patients, particularly in those with infection-associated DIC.100 Hence, TAFI may contribute (along with PAI-1) to microvascular thrombosis-induced ischemia in organs resulting in multiorgan dysfunction.
Superoxides and hydroxyl radicals are generated during sepsis and other organ injury states that predispose to DIC. Each is a proinflammatory agent that may lead to recruitment of neutrophils, formation of chemotactic factors, lipid peroxidation, and stimulation of NF-κB, which induces cytokine upregulation.101 In addition, formation of peroxynitrite by these radicals exacerbates inflammation by (1) deactivating superoxide dismutase, which ordinarily would eliminate these superoxides and other radicals, and (2) exerting damaging effects on deoxyribonucleic acid, nicotinamide adenine dinucleotide, and ATP. For example, evidence indicates that the poor response to pressors in shock-like states associated with DIC may be directly related to their deactivation by superoxides.
Adding further insult, high levels of superoxide impair vascular response to nitrous oxide, thereby creating an imbalance in the signaling to vascular cells. Because of the strategic importance of an intact endothelium for attenuating any microangiopathic process, the most devastating effect of excessive generation of superoxides and associated free radicals may be their role in inducing endothelial apoptosis, which exacerbates capillary leak.101
Vasoactive substances play a critical role in the evolution of DIC. The vasodilatory agent nitric oxide (NO) and the vasoconstrictor endothelin have been measured in experimental rat models of DIC induced by both TF infusion and lipopolysaccharide (LPS) infusion.102 LPS infusion increased both NO and endothelin remarkably, whereas TF infusion increased NO more than did LPS but did not stimulate endothelin significantly. The differential stimuli–response mechanisms may explain why LPS-induced DIC so prominently displays tissue infarction leading to multiorgan dysfunction (e.g., sepsis) compared to DIC that is predominately induced by TF exposure (e.g., head trauma).
Because there is a tight relationship between plasma lipoproteins and coagulation, it has been suggested that lipoprotein metabolism modulates coagulation in DIC.103 In vitro experiments showed that plasma large very-low-density lipoprotein, small very-low-density lipoprotein, intermediate-density lipoprotein, and low-density lipoprotein stimulate activation of coagulation by supporting factor VII activation or by stimulating monocytes to express TF.104 Lipid infusion potentiates in animals endotoxin-induced coagulation activation, as indicated by increased plasma levels of prothrombin fragments 1 and 2, thrombin–AT III complex, and PAI-1.105 High-density lipoprotein (HDL) exerts opposite effects. Administration of recombinant HDL (rHDL) ameliorates the inflammatory response, inhibits coagulation, and augments fibrinolysis,106 as reflected by reduced thrombin generation and increased levels of t-PA antigen following administration of endotoxin.
Endogenous lipid levels may have similar effects. Human subjects with low endogenous HDL-cholesterol plasma levels injected with small doses of endotoxin had a more pronounced increase in markers of coagulation activation in comparison with subjects with high endogenous HDL levels.107 Also, patients heterozygous for familial hypercholesterolemia whose low-density lipoproteins level is increased were more prone to activation of coagulation upon an inflammatory stimulus.108
Hyperglycemia and hyperinsulinemia, as seen in type 2 diabetes mellitus and the associated metabolic syndrome, affect hemostasis.109,110,111 In these circumstances, there is a marked decrease of endogenous fibrinolysis because of increased upregulation of plasma levels of PAI-1. Also, a modulatory effect of glucose/insulin on coagulation in an inflammatory setting has been described. Inflammation-induced TF gene expression was elevated in the brain, lung, kidney, heart, liver, and adipose tissues of diabetic mice compared with controls. Administration of insulin to lean mice induced enhanced inflammation-driven TF mRNA in the kidney, brain, lung, and adipose tissue.112 In a hyperglycemic normoinsulinemic study in healthy subjects, there was an increased sensitivity toward endotoxin exhibited by upregulation of TF expression.113 Strict glucose regulation in critically ill patients improves survival and reduces morbidity that is probably related to a better control of the derangement of coagulation and a faster resolution of coagulation abnormalities.103
The widespread generation of thrombin in DIC induces deposition of fibrin, which leads to the consumption of substantial amounts of platelets, fibrinogen, factors V and VIII, protein C, AT, and components of the fibrinolytic system. This situation results in massive depletion of these components that is further aggravated because of their decreased synthesis by the liver, which frequently is affected in DIC. Depending on the magnitude and nature of component depletion, bleeding, enhanced thrombosis, or both can result. Bleeding can be promoted by fibrinolysis-derived fibrin degradation products (FDPs) that exhibit anticoagulant and antiplatelet aggregation effects (see Fig. 129–1). Microangiopathic hemolytic anemia also occurs as a result of blood cells passing through vessels that are partially occluded by thrombi.
CLINICAL FEATURES
Numerous disorders can provoke DIC, but only a few constitute major causes, as can be inferred from retrospective clinical studies (Table 129–1).114 Infectious diseases and malignant disorders together account for approximately two-thirds of DIC cases in the major series (Table 129–2). Trauma was a major cause of DIC in some series, probably reflecting the specialized nature of the clinical practice in those centers.115
| Infectious diseases |
| Purpura fulminans |
| Malignancy |
Solid tumors Leukemias |
| Trauma |
Brain injury Burns |
| Liver diseases |
| Heat stroke |
| Severe allergic/toxic reactions |
| Snake bites |
| Vascular abnormalities/Hemangiomas |
Kasabach-Merritt syndrome Other vascular malformations Aortic aneurysms |
| Severe immunologic reactions (e.g., transfusion reaction) |
| Obstetrical conditions |
Abruptio placentae Amniotic fluid embolism Preeclampsia/eclampsia HELLP (hemolysis, elevated liver enzymes, and low platelet count) syndrome Sepsis during pregnancy Acute fatty liver |
| Study | Number of Patients | Infectious Disease | Trauma and Major Surgery | Malignant Disease | Liver Disease | Obstetric Complications | Miscellaneous Diseases |
|---|---|---|---|---|---|---|---|
| Minna et al.347 | 60 | 41 | 30 | 2 | 5 | 2 | 20 |
| Siegal et al.115 | 118 | 40 | 24 | 7 | 4 | 4 | 21 |
| Spero et al.122 | 346 | 26 | 19 | 24 | 8 | 0 | 23 |
| Matsuda et al.348 | 503 | 15 | 2 | 61 | 6 | 4 | 12 |
| Kobayash et al.139 | 345 | 16 | — | 55 | 4 | 5 | 20 |
| Larcan et al.349 | 361 | 15 | 14 | 6 | 3 | 38 | 24 |
Clinical manifestations are attributable to DIC, the underlying disease, or both (Table 129–3). Bleeding manifestations were common in all series of DIC cases, but considerable variation existed in the relative frequency of shock and of dysfunction of the liver, kidney, lungs, and central nervous system. These variations probably reflect the different nature of the underlying disorders in the respective series.
| Study | Number of Patients | Bleeding | Thromboembolism | Renal Failure | Liver Failure | Respiratory Failure | CNS Manifestation | Shock | Acral Cyanosis* |
|---|---|---|---|---|---|---|---|---|---|
| Minna et al.347 | 60 | 87 | 22 | 67 | NR | 78 | 65 | NR | 14 |
| Al-Mondhiry et al.116 | 89 | 76 | 23 | 39 | NR | NR | 11 | NR | 0 |
| Siegal et al.115 | 118 | 64 | 8 | 25 | 22 | 16 | 2 | 14 | 0 |
| Matsuda et al.348 | 47 | 87 | 47 | 40 | NR | 38 | NR | NR | NR |
| Spero et al.122 | 346 | 77 | NR | NR | NR | NR | NR | NR | NR |
| Larcan et al.349 | 361 | 73 | 11 | 61 | 57 | 37 | 13 | 55 | 13 |
Acute DIC frequently is heralded by hemorrhage into the skin at multiple sites.115 Petechiae, ecchymoses, and oozing from venipunctures, arterial lines, catheters, and injured tissues are common. Bleeding also may occur on mucosal surfaces. Hemorrhage may be life-threatening, with massive bleeding into the gastrointestinal tract, lungs, central nervous system, or orbit. Patients with chronic DIC usually exhibit only minor skin and mucosal bleeding.
Extensive organ dysfunction can result from microvascular thrombi or from venous and/or arterial thromboembolism (Table 129–4). For example, involvement of the skin can cause hemorrhagic bullae, acral necrosis, and gangrene. Thrombosis of major veins and arteries and pulmonary embolism occur but are rare. Cerebral embolism can complicate nonbacterial thrombotic endocarditis in patients with chronic DIC.
| Organ | Manifestation |
|---|---|
| Skin | Purpura, bleeding from injury sites, hemorrhagic bullae, focal necrosis, acral gangrene |
| Cardiovascular | Shock, acidosis, myocardial infarction, cerebrovascular events, thromboembolism in all types and caliber blood vessels |
| Renal | Acute renal insufficiency (acute tubular necrosis), oliguria, hematuria, renal cortical necrosis |
| Liver | Hepatic failure, jaundice |
| Lungs | Adult respiratory distress syndrome, hypoxemia, edema, hemorrhage |
| Gastrointestinal | Bleeding, mucosal necrosis and ulceration, intestinal ischemia |
| Central nervous system | Coma, convulsions, focal lesions, bleeding |
| Adrenals | Adrenal insufficiency (hemorrhagic necrosis) |
Both the diseases underlying DIC and the DIC itself can cause shock. For example, septicemia and excessive blood loss because of trauma or obstetric complications by themselves can cause shock. Whatever the cause of shock, its advent in cases with DIC is a serious adverse event.
Renal cortical ischemia induced by microthrombosis of afferent glomerular arterioles and acute tubular necrosis related to hypotension are the major causes of renal dysfunction in DIC. Oliguria, anuria, azotemia, and hematuria were observed in 25 to 67 percent of cases in all series (see Table 129–3).
Hepatocellular dysfunction sufficient to cause jaundice has been reported in 20 to 50 percent of patients with DIC.4,115 Infectious diseases and prolonged hypotension contribute to hepatic dysfunction.
Microthrombi, macrothrombi, emboli, and hemorrhage in the cerebral vasculature all have been held responsible for the nonspecific neurologic symptoms and signs displayed by patients with DIC.116 These manifestations include coma, delirium, transient focal neurologic symptoms, and signs of meningeal irritation. Careful exclusion of causes other than DIC is essential.
Symptoms and signs of respiratory dysfunction in DIC range from transient hypoxemia in mild cases to pulmonary hemorrhage and ARDS in severe cases.117,118,119 Pulmonary hemorrhage is heralded by hemoptysis, dyspnea, and chest pain. Physical examination reveals rales, wheezing, and occasionally a pleural friction rub. Chest imaging shows diffuse infiltration resulting from excessive intraalveolar hemorrhage. ARDS is characterized by tachypnea, auscultatory silence, hypoxemia, low lung compliance, normal wedge pressure, and “white lungs” on chest images.120 It stems from severe damage to the pulmonary vascular endothelium, which permits egress of blood components into the pulmonary interstitium and alveoli. This situation leads to intraalveolar hyaline membrane formation and severe respiratory insufficiency. ARDS can be caused by septic shock, severe trauma, fat embolism, amniotic fluid embolism, and heat stroke, all of which can also incite DIC. Yet only a fraction of patients with ARDS exhibit signs of DIC. When DIC and ARDS are simultaneously triggered, each aggravates the other. Regardless of the mechanism, ARDS is a serious complication in patients with DIC.
Both DIC and its underlying disorders contribute to the high mortality rate. Mortality correlates independently with the extent of organ dysfunction,115 the degree of hemostatic failure,121 and increasing age.122 Mortality rates in major series of patients with DIC ranged from 31 to 86 percent,121,122,123,124 whether or not heparin was administrated. Of note, there is a clear correlation between the severity of DIC and the mortality rate.121,123,124 In patients with sepsis, the presence of DIC is one of the strongest predictors of 28-day mortality.124
LABORATORY FEATURES AND DIAGNOSIS
No single laboratory test is sensitive or specific enough to allow a definite diagnosis of DIC (Table 129–5). However, some sophisticated laboratory tests, for example, thrombin–AT complex, prothrombin fragment 1.2, are sensitive to ongoing activation of coagulation pathways. Determination of soluble fibrin in plasma is one of the best parameters for detection of ongoing DIC125,126,127,128; when the concentration is above a defined threshold, a diagnosis of DIC is likely.129,130 Most of the other parameters show a sensitivity of 90 to 100 percent for the diagnosis of DIC but have a rather low specificity,131 and a wide discordance among various assays.132 FDPs may be detected by specific enzyme-linked immunosorbent assays or by latex agglutination assays, allowing rapid and bedside determination.133 None of the available assays discriminates between degradation products of crosslinked fibrin and fibrinogen, a situation that may cause spuriously high results.134,135 The specificity of high levels of FDPs is therefore limited and many other conditions, such as trauma, recent surgery, inflammation or venous thromboembolism, are associated with elevated FDPs.
| Test | Abnormality | Causes Other Than DIC Contributing to Test Result |
|---|---|---|
| Platelet count | Decreased | Sepsis, impaired production, major blood loss, hypersplenism |
| Prothrombin time | Prolonged | Vitamin K deficiency, liver failure, major blood loss |
| aPTT | Prolonged | Liver failure, heparin treatment, major blood loss |
| Fibrin degradation products | Elevated | Surgery, trauma, infection, hematoma |
| Protease inhibitors (e.g., protein C, AT, protein S) | Decreased | Liver failure, capillary leakage |
Newly developed tests are aimed at the detection of neoantigens on degraded crosslinked fibrin, one of which detects an epitope related to plasmin-degraded crosslinked γ-chain, associated with D-dimer formation. These tests better differentiate degradation of crosslinked fibrin from fibrinogen or fibrinogen degradation products.136 D-dimer level is substantially elevated in patients with DIC, but this poorly distinguishes patients with DIC from patients with venous thromboembolism, recent surgery, or inflammatory conditions.133,137
In routine practice, simple laboratory tests in conjunction with clinical considerations are used for establishing the diagnosis of DIC. The simple tests include platelet count, prothrombin time, fibrinogen level, and fibrin-related markers, such as FDP or D-dimer. Caution should be exercised when using these laboratory parameters in the algorithms described below, because an underlying disease by itself can cause an abnormality. For example, impairment of hemostasis and/or thrombocytopenia unrelated to DIC can arise from hepatic disease and from marrow involvement by leukemia. Impaired hemostasis also may occur normally in the neonatal period. Conversely, the elevated levels of some hemostatic components that are normally observed during pregnancy may obscure the presence of DIC. These limitations in laboratory diagnosis of DIC can be overcome by repeated testing, thereby following the dynamics of the process.
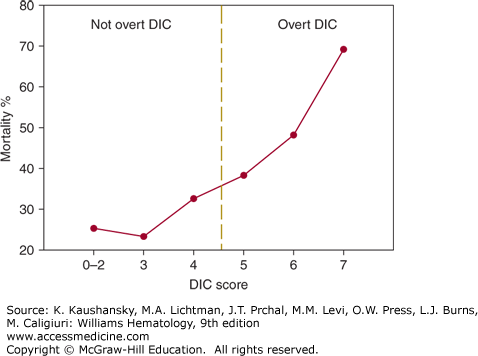
A scoring system utilizing the simple laboratory tests has been developed by the subcommittee on DIC of the International Society on Thrombosis and Haemostasis,138 and Table 129–6 summarizes a five-step diagnostic algorithm to calculate a DIC score. Tentatively, a score of 5 or more is compatible with DIC, whereas a score of less than 5 may be indicative but is not affirmative for nonovert DIC. By using receiver-operating characteristics curves, an optimal cutoff for a quantitative D-dimer assay was determined, thereby optimizing sensitivity and the negative predictive value of the system.131 Prospective studies show that the sensitivity of the DIC score is 93 percent, and the specificity is 98 percent.123 The severity of DIC according to this scoring system is related to the mortality in patients with sepsis (Fig. 129–3).124 Linking prognostic determinants from critical care measurement scores such as acute physiology and chronic health evaluation (APACHE-II) to DIC scores is an important means to assess prognosis in critically ill patients. In addition, certain biochemical indicators of organ dysfunction may imply a DIC risk. For example, serial assessment of arterial lactate has proved to be a reliable prognostic indicator of DIC development among patients with the systemic inflammatory response syndrome.139
Figure 129–3.
Number of points on the International Society of Thrombosis and Haemostasis disseminated intravascular coagulation (DIC) score and 28-day mortality in patients with severe sepsis. Data were derived from the placebo group (n = 840) in the Prowess trial on the efficacy of activated protein C in sepsis.
| 1. Presence of an underlying disorder known to be associated with DIC (see Table 129–2) | □ |
| (no = 0, yes = 2) | |
| 2. Score global coagulation test results | □ |
| Platelet count (>100 = 0; <100 = 1; <50= 2) | |
| Level of fibrin markers (soluble fibrin monomers/fibrin degradation products) | □ |
| (no increase: 0; moderate increase: 2; strong increase: 3) | |
| Prolonged prothrombin time | □ |
| (<3 s= 0; >3 s but <6 s= 1; >6 s = 2) | |
| Fibrinogen level | □ |
| (>1.0 g/L = 0; <1.0 g/L = 1) | |
| 3. Calculate score | □ |
| 4. If ≥5: compatible with overt DIC; repeat scoring daily | |
| If <5: suggestive (not affirmative) for nonovert DIC; repeat next 1–2 days |
Criteria for less-overt DIC have been more difficult to establish.138,140 In the algorithm for nonovert DIC, the global coagulation tests are scored as with the overt DIC algorithm; however, when scoring by the algorithm is being serially repeated, improvement in any laboratory test confers a negative score (rather than a zero or neutral score). This “trend” scoring allows longitudinal assessment of the patient’s microangiopathy and, when therapy has been instituted, inference on whether the therapy has improved the course of the disease.121,141 Measurements of several markers for assessing the risk of progression from nonovert to overt DIC and prediction of multiorgan dysfunction are potentially valuable and in the future can be accommodated in the nonovert DIC score. For example, impaired fibrinolysis may play a particularly important role in multiorgan dysfunction resulting from DIC of sepsis.142 Therefore, assaying PAI-1, plasmin–antiplasmin complexes, or TAFI in septic patients may be important. Another highly sensitive early marker of impending DIC is a monoclonal antibody against APC that identifies a calcium ion-dependent epitope involved in factor Va inactivation.143 Whether serial measurement of von Willebrand factor-cleaving protease also will identify individuals at risk early in their disease course, or will help differentiate individuals with microangiopathy who are not prone to progress, needs further data.144,145
Techniques, such as rotational thrombelastography (ROTEM) enable bedside performance of this test and has again become popular recently in acute care settings.146 The theoretical advantage of thrombelastography (TEG) over conventional coagulation assays is that is provides an idea of platelet function as well as fibrinolytic activity. Hyper- and hypocoagulability as demonstrated with TEG was shown to correlate with clinically relevant morbidity and mortality in several studies, although its superiority over conventional tests has not unequivocally been established.147 Even though there are no systematic studies on the diagnostic accuracy of TEG for the diagnosis of DIC, the test may be useful for assessing the global status of the coagulation system in critically ill patients.148
SPECIFIC UNDERLYING DISORDERS
Bacterial infections are among the most common causes of DIC.5,149 Certain patients are particularly vulnerable to infection-induced DIC, such as immune-compromised hosts, asplenic patients whose ability to clear bacteria, particularly pneumococci and meningococci, is impaired, and newborns whose coagulation inhibitory systems are immature. Infections are frequently superimposed on trauma and malignancies, which themselves are potential triggers of DIC. In addition, infections can aggravate bleeding and thrombosis by directly inducing thrombocytopenia, hepatic dysfunction, and shock associated with diminished blood flow in the microcirculation.150 Clinically overt DIC may occur in 30 to 50 percent of patients with Gram-negative sepsis.151,152 DIC is similarly common in patients with Gram-positive sepsis.153,154 Extreme examples of sepsis-related DIC are (1) group A streptococcus toxic shock syndrome, characterized by deep tissue infection, vascular collapse, vascular leakage, and multiorgan dysfunction; a streptococcal M protein forms complexes with fibrinogen, and these complexes bind to β2 integrins of neutrophils leading to their activation155
Related posts:
 Hematopoietic Stem Cells, Progenitors, and Cytokines
Examination of the Marrow
Therapeutic Apheresis: Indications, Efficacy, and Complications
Hemolytic Anemia Resulting from Infections with Microorganisms
Structure and Composition of the Erythrocyte
Disorders of Hemoglobin Structure: Sickle Cell Anemia and Related Abnormalities
Hematopoietic Stem Cells, Progenitors, and Cytokines
Examination of the Marrow
Therapeutic Apheresis: Indications, Efficacy, and Complications
Hemolytic Anemia Resulting from Infections with Microorganisms
Structure and Composition of the Erythrocyte
Disorders of Hemoglobin Structure: Sickle Cell Anemia and Related Abnormalities
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


