- Patients with type 1 diabetes classically present early, require continuous insulin treatment and carry autoimmune markers such as antibodies to glutamic acid decarboxylase (GAD).
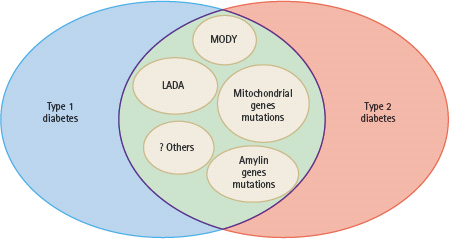
- There are other types of diabetes with type 1 phenotype secondary to heterogeneous etiologies including: maturity-onset diabetes of the young (MODY) and other forms of monogenic diabetes caused by mutations of mitochondria, amylin or pathways implicated in pancreatic β-cell function; latent autoimmune diabetes of adults (LADA); fulminant type 1 diabetes presenting with diabetic ketoacidosis after viral infections and another form which reverts to a clinical course resembling type 2 diabetes after the initial ketotic presentation.
- The correct diagnosis of these disorders with type 1 phenotype is clinically important because of their different clinical course, prognosis and management.
Introduction
Classic type 1 diabetes mellitus (T1DM) is considered an autoimmune disease with pancreatic β-cell destruction. Affected subjects typically have onset of their disease at a young age with an acute presentation including diabetic ketoacidosis requiring continuous insulin treatment [1]. With a better understanding of the epidemiology and molecular mechanism of diabetes, however, clinical features such as the younger age of onset (e.g. less than 35 years old) or dependence on insulin treatment cannot adequately define the etiology of patients presenting with hyperglycemia.
Furthermore, there are major ethnic differences in disease pattern in terms of presentation and natural progression. In Caucasians, over 90% of patients with diabetes diagnosed before the age of 35 years have type 1 disease [1]. By contrast, autoimmune T1DM is uncommon in non-Caucasian populations [2–5]. Using Hong Kong as an example, which has a relatively homogenous southern Chinese population leading an affluent lifestyle, less than 10% of adults presenting with diabetic ketoacidosis have autoimmune markers. Similarly, only 10% of Hong Kong Chinese patients with young onset of disease had a type 1 presentation or antibodies to glutamic acid decarboxylase (GAD) [6]. Similar epidemiologic findings have also been reported in other Asian populations from India, Malaysia, Singapore and Mainland China [7]. In most case series, 60–80% of Caucasian people with T1DM have autoimmune markers such as autoantibodies to GAD and/or islet cell antigens (e.g. ICA-512). Conversely, 5–20% of young Asian patients with a non-ketotic presentation have autoimmune markers with a wide range of insulin reserve. These findings suggest that latent autoimmune diabetes in adults (LADA) is not uncommon in young patients with diabetes, especially those of Asian ethnicity, and there is considerable overlap between type 1 and 2 diabetes phenotypes (Figure 9.4) [6,8].
Figure 9.4 There are considerable overlaps diabetes between the phenotypes of type 1 and type 2 diabetes because of the heterogeneous genetic and autoimmune etiologies. These include maturity-onset diabetes of the young (MODY), latent autoimmune diabetes in adult (LADA) and genetic variants affecting the insulin, amylin and mitochondrial pathways. Other rare causes include fibrocalculous pancreatic diabetes and fulminant type 1 diabetes.
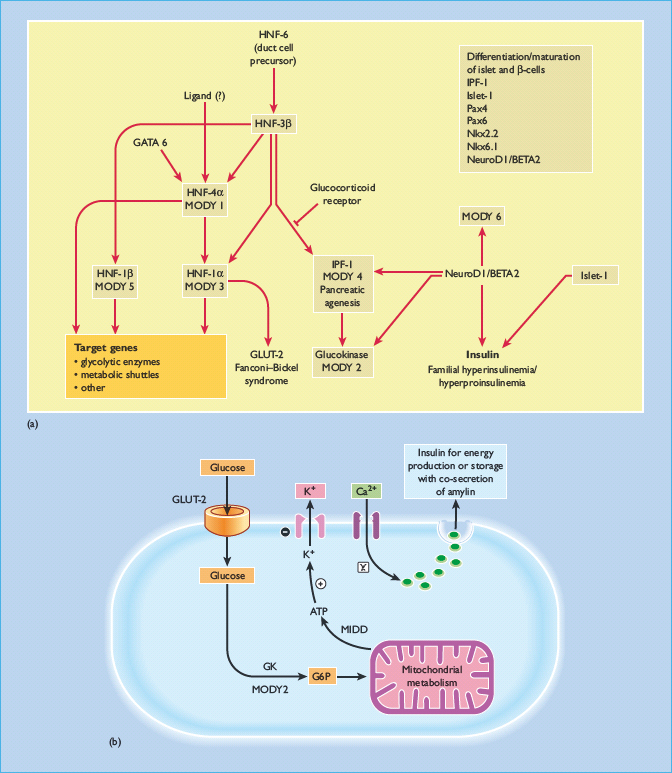
Our current understanding of the molecular pathways involved in the neogenesis, differentiation and maturation of pancreatic β-cells as well as the intracellular signaling mechanisms leading to insulin secretion are summarized in Figure 9.5. This large body of knowledge has provided the basis for the discovery and description of subtypes of diabetes with predominant β-cell failure from causes other than autoimmunity, such as monogenic diabetes. Patients with monogenic diabetes often have young onset of disease and lean body mass (see Chapter 15). They may also have delayed presentation with complications brought about by the insidious nature of their symptoms [9,10]. With the rising prevalence of young onset diabetes especially in low and middle income countries [11], there is a need for health care providers to be aware of these non-classic presentations of T1DM, because they have important implications on clinical management and family screening.
Figure 9.5 (a) The cascade of transcription factors involved in pancreatic development as well as neogenesis, differentiation and maturation of pancreatic β-cells. Maturity-onset diabetes of the young (MODY) includes subtypes with mutations in transcription factors, namely MODY 1 with mutations of hepatic nuclear factor (HNF-4α); MODY 3:HNF-1α; MODY 4:insulin promotion factor (IPF-1); MODY 5:HNF-1β; MODY 6: NeuroDI: neurogenic differentiation 1 and MODY 7: carboxyl ester lipase (CEL) as well as glucokinase (GK) which is the glucose-sensor of the pancreatic β-cells (MODY 2). There are also interactions of glucose transporter 2 (GLUT 2) and endodermal factor, including GATA and various important genes and transcription factors governing the differentiation and maturation of pancreatic islet and beta cells. These include Pax genes family and genes encoding the homeodomain transcription factor Nkx 2.2 and Nkx 6.1. In South Asian Indian population, interaction of the NeuroD1, neurogenin-3 (NEUROG3) and HNF-1α genes has been observed to have combined effect in controlling islet cell development and insulin secretion, thus contributing to the overall glucose tolerance [93]. (b) The multiple steps involved in regulation of insulin secretion commencing with sensing of ambient blood glucose level by GLUT-2, glycolysis by GK, and ATP production by mitochondria. The generated ATP particles then close the potassium channel leading to membrane depolarization and opening of calcium channels. The intracellular calcium influx is associated with translocation of insulin and amylin containing vesicles to the cellular surface for extracytosis. During these processes, transcription factors are also activated resulting in insulin gene transcription and production to replenish the insulin containing vesicles and maintain continuous insulin secretion. GLUT, glucose transporter; MIDD, maternal inherited diabetes and deafness; MODY, maturity-onset diabetes of the young.
Atypical diabetes: heterogeneous etiologies of young-onset diabetes
In 1980, Winter et al. [12] first described a cohort of 129 African-American youths with acute ketotic presentation, of whom 12 patients subsequently did not require insulin and followed a clinical course resembling type 2 diabetes (T2DM). Since then, a number of reports have shown that there is a poor correlation between the mode of presentation of hyperglycemia, clinical course, need for insulin treatment and autoimmune status in different ethnic groups including Asians [13–15]. Many of these patients did not have HLA genotypes or autoantibodies typical of autoimmune T1DM. Some patients were obese, with insulin resistance and glucotoxicity contributing to the initial ketotic presentation [16].
Monogenic diabetes caused by genetic mutations encoding pathways implicated in insulin synthesis and secretion may lead to young-onset diabetes with atypical presentation. In young Chinese patients with diabetes, there is a higher prevalence of parental history of diabetes (32–47%), particularly maternal history, compared with those having a late onset of disease (12–19%) [10,17,18]. A progressively earlier age of onset of disease in successive generations has also been reported in some of these affected families [19]. These patients may exhibit a mixed phenotype, including young age of diagnosis, insulinopenia and normal body weight (as in type 1), with a non-ketotic state typical of T2DM, despite lack of insulin resistance and metabolic syndrome [20].
Monogenic diabetes
Maturity-onset diabetes of the young
Patients with maturity-onset diabetes of the young (MODY) typically present before the age of 25 years with a strong family history suggestive of autosomal dominant inheritance (see Chapter 15). Despite a non-ketotic mode of presentation, these patients often have features of abnormal pancreatic β-cell function. Some patients with MODY have a rapid deterioration in glycemic control after initial presentation while others experience mild hyperglycemia and do not require insulin despite having a long duration of disease.
To date, several subtypes of MODY have been reported. These include mutations of transcription factors: MODY 1: hepatic nuclear factor 4α (HNF-4α); MODY3; HNF-1α or transcription factor 1 (TCF1); MODY 4: insulin promotion factor 1 (IPF-1); MODY 5: HNF-1β or transcription factor 2 (TCF-2); MODY 6: neurogenic differentiation 1 (NeuroD1) and MODY 7: carboxyl ester lipase (CEL) and glucokinase which is the glucose-sensor of the pancreatic β-cells (MODY2). Transcription factors have key roles in pancreatic development including differentiation and proliferation of β-cells (Figure 9.4b). While genetic mutations in transcription factors typically cause significant insulin insufficiency and hyperglycemia, common polymorphisms of some of these transcription factors (e.g. HNF-1α [21], HNF-4α [22] and HNF-1β [23]) have also been shown to be associated with increased risk of diabetes or metabolic traits which may interact with other genetic or environmental and/or lifestyle factors to give rise to overt diabetes.
Over 80% of Caucasian patients with classic MODY (i.e. age of onset less than 25 years with autosomal pattern of inheritance) have been reported to have mutations in HNF-1α or glucokinase, while other MODY subtypes (HNF-4α, HNF-1β and IPF-1 mutations) were less common [24]. The frequencies of HNF-1α mutations ranged 25–50% in French [25], 36% in German [26], 13–18% in British [27], 8% in Japanese [28] and 5% in Chinese patients [9]. In unrelated young Chinese patients with diabetes, 5–10% were found to have glucokinase or HNF-1α mutations [13,29,30].
Although patients with some forms of MODY (e.g. MODY 2) have mild clinical course and rarely develop complications, other forms of MODY may be associated with severe insulin insufficiency and complications often brought about by the late presentation and/or delayed use of insulin [10]. In light of the potential long duration of disease, young patients with diabetes are more prone to develop microvascular complications than those with late onset of disease [31], thus emphasizing the importance of family screening.
Furthermore, heterogeneous mutations in these transcription factors can be expressed sequentially in renal tubules with possible roles in various stages of the development of renal tubules [32]. Thus, patients with MODY may have heterogeneous phenotypes with metabolic and renal manifestations. Patients with MODY 3 from mutations in HNF-1α were found to have a low renal threshold for glucose [33] while patients with MODY 5 from mutations in HNF-1β may have mild diabetes but increased susceptibility to severe renal disease and other urogenital malformations [34]. In a consecutive unrelated cohort consisting of 74 young Chinese patients with T2DM and nephropathy, a novel missense genetic variant in exon 3 (E260D, GAG→GAC) of the HNF-1β gene was identified. Extended family analysis revealed four other siblings carrying this variant with heterogeneity in clinical presentation that included one member with uncomplicated diabetes, one with impaired glucose tolerance and one with microalbuminuria with normal glucose tolerance. A silent polymorphism Q378Q was identified in another unrelated subject in this study. While these findings will need replication in independent and larger cohorts, the phenotypic heterogeneity associated with these genetic variants is noteworthy [35].
Mitochondrial gene mutations
Mitochondria are important intracellular organelles in the maintenance of glucose homeostasis and energy balance. Mitochondria have their own genome and unlike nuclear DNA which is protected by histones, mitochondrial DNA is more vulnerable to oxidative stress and environmental toxins. Superoxide radicals generated by the mitochondrial respiratory chain are a major source of damage to mitochondrial DNA. Aged patients with a positive family history of diabetes have a high frequency of mitochondrial mutations [36]. Because of its maternal inheritance, mitochondrial DNA is a well-known cause of a subtype of maternally inherited diabetes mellitus [37].
In 1992, an A3243G mutation in the mitochondrial DNA coding for tRNALeu(UUR) (mt3243) was first reported. This form of diabetes was found in patients with both type 1 and type 2 diabetes and was characterized by maternal inheritance and deafness [38]. In a random cohort of Chinese patients with diabetes, 1–3% had this mutation with either type 1 or type 2 clinical course [39–41]. Other point mutations associated with increased risk of diabetes include sites at 3316, 3394 and 14577 as well as deletion and rearrangement in mitochondrial DNA [36].
In keeping with its candidacy as a “thrifty gene,” the frequency of a common polymorphism of the mitochondrial DNA (T16189C) is higher in Chinese subjects with metabolic syndrome than those without (44% vs 33%), after adjustment for age and body mass index (BMI) [42]. In a meta-analysis, Asian subjects without diabetes had a higher frequency of the 16189C variant than their European counterparts (31.0% vs 9.2%) [43]. Despite negative reports in European populations [43], there are consistent data showing the risk association of the 16189C variant with T2DM in Asians [44,45].
Amylin gene mutations
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


