DISORDERS OF SEXUAL DIFFERENTIATION
No classification scheme for the various disorders of sexual differentiation is ideal. The easiest approach is perhaps to consider such disorders on the basis of whether they affect primarily the gonads, the ductal structures involving the gonads, or the external genitalia (Table 90-2).
 |
ERRORS IN GONADAL DEVELOPMENT
TRUE HERMAPHRODITISM
By definition, true hermaphrodites possess testicular and ovarian tissue, either separately or, more commonly, together as one or more ovotestes.49a Most true hermaphrodites (50–70%) have a 46,XX chromosomal complement; however, some have 46,XX/46,XY, 46,XX/47,XXY, 46,XY, or other complements.50 Some evidence suggests that phenotype reflects karyotype, but this is unproved.50
Phenotype.
Approximately two-thirds of true hermaphrodites are raised as males, although their external genitalia may be frankly ambiguous or predominantly female. Paradoxically, breast development usually occurs at puberty despite the presence of male external genitalia; virilization does not.
Gonadal tissue may be located in the ovarian, inguinal, or labioscrotal region. A testis or an ovotestis is more likely to be present on the right side than on the left. The greater the proportion of testicular tissue in an ovotestis, the greater the likelihood of gonadal descent. In 80% of ovotestes, the testicular and ovarian components exist immediately adjacent to each other in end-to-end fashion.51,52 An ovotestes usually can be detected by inspection, or possibly by palpation, because testicular tissue is softer and darker than ovarian tissue. Spermatozoa are rarely present; however, apparently normal oocytes often are present, even in ovotestes.
Usually, a uterus is present, and commonly it is bicornuate or unicornuate. The absence of a uterine horn usually indicates the presence of an ipsilateral testis or ovotestis. The fimbriated end of the fallopian tube may be occluded ipsilateral to an ovotestis, and squamous metaplasia of the endocervix may occur. Most true hermaphrodites with a uterus menstruate. Five 46,XX true hermaphrodites have become pregnant—usually, but not always, after the removal of testicular tissue.53,54
Typically, the diagnosis of true hermaphroditism is made only after excluding the more common forms of male and female pseudohermaphroditism. A male sex-of-rearing is possible if genital status permits reconstruction and if the inappropriate (i.e., ovarian) tissue is extirpated.
All gonadal tissue should be biopsied, and tissue not compatible with the gender assignment removed. Despite the “dysgenetic” nature of the gonads in true hermaphrodites and the fact that a Y chromosome is sometimes present, gonado-blastomas have been reported far less frequently than in XY gonadal dysgenesis.55,56 Carcinoma of the breast also has been reported.
Etiology.
The cause of true hermaphroditism is uncertain but heterogeneous. Cases with 46,XX/46,XY karyotypes presumably
result from chimerism, the presence of two or more cell lines, each derived from different zygotes, in a single individual. In one survey, 6 of 28 46,XX/46,XY cases were proved chimeras.50 Experimental production of XX/XY mouse chimeras usually does not result in true hermaphroditism, however, nor do 46,XX/46,XY humans always have true hermaphroditism. The 46,XX/47,XXY cases may result from chimerism or mitotic nondisjunction.
result from chimerism, the presence of two or more cell lines, each derived from different zygotes, in a single individual. In one survey, 6 of 28 46,XX/46,XY cases were proved chimeras.50 Experimental production of XX/XY mouse chimeras usually does not result in true hermaphroditism, however, nor do 46,XX/46,XY humans always have true hermaphroditism. The 46,XX/47,XXY cases may result from chimerism or mitotic nondisjunction.
A few 46,XX true hermaphrodites doubtless result from undetected chimerism; however, for various reasons, undetected chimerism cannot easily explain all 46,XX true hermaphrodites. The presence of testicular tissue in 46,XX individuals is perplexing, because the TDF is localized to the Y chromosome, specifically the short arm. Plausible explanations for the presence of testes in individuals who ostensibly lack a Y chromosome include translocation of TDF from the Y to an X, translocation of TDF from the Y to an autosome, undetected mosaicism or chimerism, and activation of sex-reversal genes, perhaps located on autosomes. The first or second hypothesis is supported by the detection of H-Y antigen in almost all 46,XX true hermaphrodites.19 Especially impressive are observations of HY antigen in each of two 46,XX true hermaphrodite sibs.57 However, cloned DNA sequences known to be near TDF have not been detected in true hermaphrodites.58 These observations contrast with those in 46,XX males, suggesting different pathogenetic mechanisms for the two sex-reversal states. Aggregates of sibs of 46,XX true hermaphrodites suggest that recessive genetic factors, perhaps activation of autosomal loci ordinarily suppressed in 46,XX individuals, are responsible.59,60 That 46,XX males and 46,XX true hermaphrodites have occurred in multiple generations of the same kindreds may or may not be attributable to the same mechanism.61,62 That XX males in some of these kindreds have not, in contrast to the usual male, shown TDF favors perturbation of an autosomal region as the cause of XX true hermaphroditism.34,35,63,64 Indeed, SRY is almost always present.
GONADAL DYSGENESIS
In ovarian dysgenesis, the gonads traditionally are said to exist in the form of streak gonads (Fig. 90-14). The streaks are devoid of germ cells, accounting for the term gonadal dysgenesis. The streak is located in the position ordinarily occupied by the ovary. In the small percentage of individuals with gonadal dysgenesis who survive until adulthood, the normal gonad is usually replaced by a white fibrous streak 2 to 3 cm long and ˜0.5 cm wide. This streak gonad is characterized histologically by interlacing waves of dense fibrous stroma, devoid of oocytes but otherwise indistinguishable from normal ovarian stroma. The karyotypes of affected individuals differ widely, with gonadal dysgenesis observed in a spectrum of karyotypes, including 45,X; 46,XX; 46,XY; various mosaics; and X or Y structural aberrations.
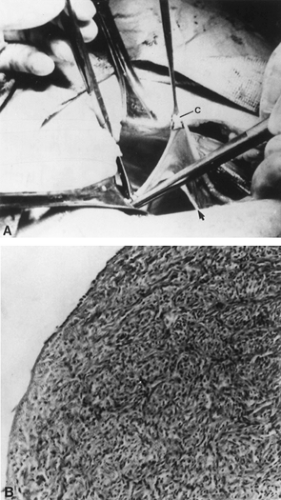 FIGURE 90-14. A, Streak gonad of patient with XY gonadal dysgenesis. Clamp (C) is elevating a fallopian tube. White streak perpendicular to forceps at lower right is the gonad (arrow). A streak gonad from a 45,X individual would look identical. B, Histologic appearance of streak gonad from an individual with 45,X gonadal dysgenesis. Only fibrous stroma is present; no oocytes are obvious. (From Simpson JL. Disorders of sexual differentiation: etiology and clinical delineation. New York: Academic Press, 1976.) |
Turner syndrome is the term most frequently applied to the picture seen in individuals with gonadal dysgenesis. This designation is confusing, however, and application of the term gonadal dysgenesis in any instance of streak gonads is perhaps more appropriate. The term Turner stigmata is reserved for short stature and certain other somatic anomalies.7
45,X Embryonic Lethality and Its Cellular Explanation.
Approximately 50% of spontaneous abortions occurring during the first 3 months of gestation are associated with a chromosomal abnormality. Twenty percent of chromosomally abnormal abortuses show monosomy X.65 Of all pregnancies, 12% to 15% or more terminate in first-trimester spontaneous abortions, and 1% to 2% of all conceptions must be 45,X. Because the incidence of 45,X at birth is only ˜1 per 10,000 females, >99% of all 45,X embryos can be assumed to be aborted. The high rate at which 45,X cell lines are spuriously detected in chorionic villi, however, raises the possibility that the frequency of 45,X in embryos is overestimated. Monosomy X may be relatively restricted to villi or membranes, those tissues most often available for cytogenetic studies, and the aborted embryo may have a different chromosomal complement. In any case, intrauterine growth restriction is characteristic of the rare surviving 45,X neonate.66
GONADAL DEVELOPMENT AND X-CHROMOSOME INACTIVATION. The absence of oocytes in monosomy X is the result of increased oocyte atresia, not failure of germ-cell formation. The 45,X embryos and 45,X neonates have germ cells.29,30 Inasmuch as germ cells are present in 45,X embryos, the fact that 3% to 5% of 45,X individuals menstruate spontaneously is not too surprising. Several fertile 45,X individuals have been reported.67
Because (according to the Lyon hypothesis) X chromosomes in excess of one are inactivated, why 45,X individuals should manifest developmental abnormalities is not so obvious. Relatively normal ovarian development occurs in 39,X mice and in most other monosomy X mammalian organisms. Data support several related explanations for these findings. First, some loci on the human heterochromatic (inactive) X are not inactivated. For example, the locus for steroid sulfatase, which is located on Xp, is not inactivated, and it would not be surprising if ovarian
determinants likewise escaped X inactivation. Second, X inactivation never occurs in human oocytes, as evidenced by the fact that females who are heterozygous for the enzyme glucose-6-phosphate dehydrogenase synthesize both alleles in oocytes.68 The parental origin of 45,X is of interest. In humans, 70% of live-born 45,X individuals have lost a paternal sex chromosome.69 This helps explain why mean maternal age is not increased for 45,X abortuses or live borns.70 Murine monosomy (39,X) also results from the loss of a paternal sex chromosome at the time of fertilization.71
determinants likewise escaped X inactivation. Second, X inactivation never occurs in human oocytes, as evidenced by the fact that females who are heterozygous for the enzyme glucose-6-phosphate dehydrogenase synthesize both alleles in oocytes.68 The parental origin of 45,X is of interest. In humans, 70% of live-born 45,X individuals have lost a paternal sex chromosome.69 This helps explain why mean maternal age is not increased for 45,X abortuses or live borns.70 Murine monosomy (39,X) also results from the loss of a paternal sex chromosome at the time of fertilization.71
Most commonly, individuals with gonadal dysgenesis have a 45,X karyotype and present with the Turner stigmata.72 Secondary sexual development usually does not occur in 45,X individuals (Fig. 90-15). Pubic and axillary hair fail to develop in normal quantity. Although well differentiated, the external genitalia, the vagina, and the müllerian derivatives (e.g., uterus) remain small. As is true for virtually all individuals with gonadal dysgenesis, estrogen and androgen levels are decreased; levels of follicle-stimulating hormone (FSH) and luteinizing hormone (LH) are increased.
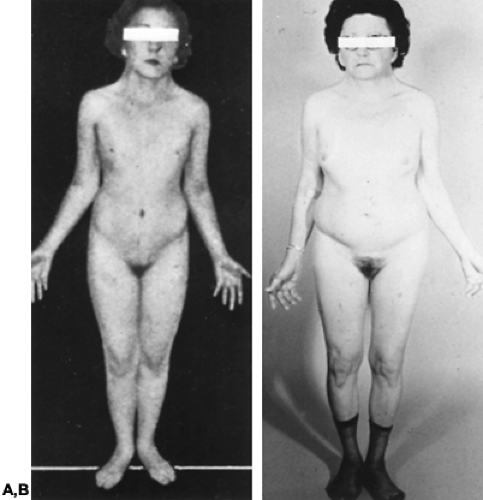 FIGURE 90-15. A, Appearance of one of original seven cases of Turner syndrome reported in 1938. B, Same patient, photographed in 1972, almost 35 years after publication of the original case report. The karyotype was documented to be 45,X. The patient had received little estrogen in intervening years and experienced severe osteoporosis. (Courtesy of Dr. R. Rebar and Dr. S. S. C. Yen, University of California, San Diego.) |
NONGONADAL FEATURES OF TURNER SYNDROME. The common anomalies of Turner syndrome include epicanthal folds, high arched palate, low nuchal hairline, webbed neck, shield like chest, coarctation of the aorta, ventricular septal defect, renal anomalies, pigmented nevi, lymphedema, hypoplastic nails, and cubitus valgus (Table 90-3). Inverted nipples and double eyelashes may be present as well. No feature is pathognomonic, but in aggregate, they form a spectrum of anomalies more likely to exist in 45,X individuals than in normal 46,XX individuals. These anomalies are the Turner stigmata, the presence of which suggests the coexistence of gonadal dysgenesis.
 |
Individuals with a 45,X karyotype have low birth weights (adjusted mean, 2851.1 ± 65.1 g).66 Total body length at birth is sometimes less than normal, but often it is normal. Height velocity before puberty generally lies in the 10th to the 15th percentile, and the mean heights of 45,X adults (16 years or older) range from 141 to 146 cm (55.5–57.5 in).72,73,74 and 75 In untreated patients, the epiphyses remain open; additional growth occurs when sex steroids are administered. Despite the diminished final height of such patients, their adult stature tends to correlate with parental height.75 (Also see ref. 75a.)
That not all patients with gonadal dysgenesis are short indicates that sex steroid deficiency is not the cause. For example, normal stature is characteristic of individuals with 46,XX gonadal dysgenesis. Growth hormone (GH) levels have long been considered essentially normal in individuals with gonadal dysgenesis.76,77 Cellular resistance to GH, however, has been suggested. This relative resistance may be overcome by treatment with exogenous GH at higher doses than those used for classic GH deficiency.78 Anti-GH antibodies have been detected, and GH reserve may be decreased.79,80 More evidence suggests that abnormalities in GH or insulin-like growth factor-I (IGF-I), which fall below the normal range after 8 years of age, are secondary to the lack of gonadal activation and estrogen secretion.81
Short stature may be common in gonadal dysgenesis because the epiphyses are structurally abnormal. This hypothesis is compatible with observations that decreased growth occurs in the long bones, teeth, and skull.82,83 One aspect of Turner syndrome may be a skeletal dysplasia.
Most 45,X patients have normal intelligence, but any given 45,X patient has a slightly higher probability of being retarded than a 46,XX individual. The frequency of overt retardation is 11% to 17%.72 Biases of ascertainment dictate that this prevalence probably represents the maximum risk. Performance IQ is definitely lower than verbal IQ, however; 45,X individuals have an unusual cognitive defect characterized by an inability to appreciate the shapes and relations of objects with respect to one another (i.e., space-form blindness).84,85 and 86 The patients usually appear socially immature, probably in part because they are short and sexually immature.87
METABOLIC ALTERATIONS. Diabetes mellitus, thyroid disease, and essential hypertension often are present in individuals with 45,X karyotypes. Abnormal oral glucose-tolerance tests may occur in as many as 40% of these individuals.88 Both autoimmune thyroiditis and Graves disease are observed with increased frequency in patients with Turner stigmata. Approximately one-third of adult 45,X patients have essential hypertension, which also may occur in young 45,X girls. Therapy for any metabolic alterations is not unique, although reduction or careful monitoring of exogenous estrogen therapy may be necessary.
Abnormalities of the X Chromosome.
Several different abnormalities of the X chromosome have been associated with gonadal dysgenesis. The cytologic origin of these defects was considered earlier.
DELETION OF THE SHORT ARM OF CHROMOSOME X. A terminal deletion of the X short arm [del(Xp)] may or may not cause gonadal dysgenesis, short stature, and other features of the Turner stigmata. The phenotype depends on the amount of Xp that is deficient.
Spontaneous menstruation, albeit usually leading to secondary amenorrhea, has occurred in almost 40% of reported 46,X,del(X)(p11) individuals (see Fig. 90-7). Almost all 46,X,del(X)(p21) individuals menstruate, but only approximately one-half become pregnant.7,34,89,90 These data indicate that ovarian tissue persists more often in del(Xp) individuals than in 45,X individuals and that complete ovarian failure (with primary amenorrhea) occurs only if the proximal and the terminal portions of Xp are deleted. However, the mean adult heights are 140 cm (55 in) for 46,X,del(X)(p11) and 146.5 cm (57.5 in) for 46,X,del(X)(p21) persons89,91 (see Fig. 90-8). Inasmuch as 46,X,del(X)(p21) individuals are short but have normal ovarian function, determinants for ovarian maintenance and for stature must be located in different regions of Xp; statural determinants are more distal.35,36,91 No evidence yet exists that any given X-specific probe bears a special relation to X-ovarian or X-structural determinants.
ISOCHROMOSOME FOR THE X LONG ARM (1[XQ]). Almost all 46,X,i(Xq) patients have streak gonads, short stature, and features of the Turner stigmata. In addition to having a duplication of Xq,i(Xq), these individuals differ from del(X)(p11) persons because the terminal portion and almost all of Xp is deleted. The better gonadal function in del(X)(p11) than in i(Xq) individuals is consistent with the location of ovarian determinants at several different sites on Xp. A locus on Xp may be deleted in i(Xq) karyotype but be retained near the centromere in 46,X,del(X)(p11) cases. Because duplication of Xq (i.e., 46,X,i[Xq]) does not compensate for deficiency of Xp, the gonadal determinants on Xq and Xp must have different functions. Whether duplication of Xq per se produces abnormalities is unknown.
DELETION OF THE LONG ARM OF CHROMOSOME X (DEL[XQ]). Most patients with a deletion of the X long arm have streak gonads and never menstruate. This is especially true of individuals with del(X)(q13). However, deletions of distal Xq are more likely to be associated with premature ovarian failure than with primary amenorrhea. The Xq appears to contain more than one region that is required for normal ovarian function. Perhaps several Xq loci can affect ovarian function in additive fashion. The only clue to the nature of any of these products is the suggestion that the diaphanous (DIA) gene, localized to Xq25, plays a role; however, other genes in distal Xq are also pivotal to ovarian development.
Mosaicism
MOSAICISM INVOLVING ONLY X CHROMOSOMES. The 45,X/46,XX individuals have fewer anomalies than 45,X individuals. In one survey, 12% of 45,X/46,XX individuals menstruated, compared with only 3% of 45,X individuals.72 In that survey, the mean adult height was greater in 45,X/46,XX persons than in 45,X individuals. More mosaic patients (25%) than nonmosaic patients (5%) reach adult heights greater than 152 cm (60 in). Somatic anomalies are less likely to occur in 45,X/46,XX than in 45,X patients.
MOSAICISM WITH A Y CHROMOSOME. Individuals with a 45,X cell line and at least one line containing a Y chromosome manifest a variety of phenotypes, ranging from almost normal males with cryptorchidism or penile hypospadias to females indistinguishable from those with the 45,X Turner syndrome. The different phenotypes presumably reflect different tissue distributions of the various cell lines, although this assumption remains unproven. At any rate, 45,X/46,XY individuals may show unambiguous female external genitalia, ambiguous external genitalia, or almost normal male external genitalia.
Some 45,X/46,XY individuals with female external genitalia have the Turner stigmata and are clinically indistinguishable from 45,X individuals. Others, however, are female but of normal stature and without somatic anomalies. As in other types of gonadal dysgenesis, the external genitalia, vagina, and müllerian derivatives remain unstimulated because of deficient sex steroids. Breasts fail to develop, and little pubic or axillary hair grows. In fact, breast development in a 45,X/46,XY individual should lead one to suspect an estrogen-secreting tumor, most commonly a gonadoblastoma or dysgerminoma.
The streak gonads of 45,X/46,XY individuals usually are indistinguishable histologically from the streak gonads of individuals with 45,X gonadal dysgenesis. However, gonadoblastomas or dysgerminomas develop in 15% to 20% of 45,X/46,XY individuals.55,56,94 Such neoplasms may arise as early as the first two decades of life. Gonadoblastomas occur almost exclusively in 46,XY or 45,X/46,XY individuals and usually are benign. However, they may be associated with dysgerminomas or other germ-cell tumors that are malignant. The gonads of 45,X/46,XY individuals should be extirpated regardless of the patient’s age. Because of the risk of neoplasia, 45,X/46,XY gonadal dysgenesis should be differentiated from forms of gonadal dysgenesis lacking a Y chromosome.
When the polymerase chain reaction for SRY was used, unrecognized Y-chromosome material was found in 1 of 40 patients with Turner syndrome.95 Because the detection of SRY sequences in patients with gonadal dysgenesis is correlated with the presence of Y-chromosomal DNA and carries the risk of tumor development, the application of this technique in search of SRY may be justified in all individuals with this disorder.
Individuals may show one streak gonad and one dysgenetic testis. The terms asymmetric gonadal dysgenesis or mixed gonadal dysgenesis are often applied to such individuals. They usually have ambiguous external genitalia. Many investigators believe that the phenotype of asymmetric gonadal dysgenesis is almost always associated with 45,X/46,XY mosaicism, with ostensibly nonmosaic cases reflecting merely an inability to analyze appropriate tissues. Most 45,X/46,XY individuals with ambiguous
external genitalia have müllerian derivatives (e.g., a uterus). The presence of a uterus is helpful diagnostically, because a uterus is absent in most genetic forms of male pseudohermaphroditism. If an individual has ambiguous external genitalia, bilateral testes, and a uterus, one may reasonably infer the presence of 45,X/46,XY mosaicism, whether or not both lines can be demonstrated cytogenetically. Occasionally, the uterus is rudimentary, or a fallopian tube fails to develop ipsilateral to a testis.
external genitalia have müllerian derivatives (e.g., a uterus). The presence of a uterus is helpful diagnostically, because a uterus is absent in most genetic forms of male pseudohermaphroditism. If an individual has ambiguous external genitalia, bilateral testes, and a uterus, one may reasonably infer the presence of 45,X/46,XY mosaicism, whether or not both lines can be demonstrated cytogenetically. Occasionally, the uterus is rudimentary, or a fallopian tube fails to develop ipsilateral to a testis.
The 45,X/46,XY mosaicism has less commonly been detected in individuals with almost normal male external genitalia. In some individuals, hypospadias is present, but the sex-of-rearing is unequivocally male.
The 45,X/47,XXY and 45,X/46,XY/47,XXY complements exist, albeit much less often than 45,X/46,XY. These complements are associated with the same phenotypic spectrum as 45,X/46,XY. Of particular interest is one family in which two and possibly three sibs had 45,X/46,XY/47,XYY mosaicism.96 The parents were second cousins, suggesting recessive factors.
Gonadal Dysgenesis in 46,XX Individuals.
The first individual with gonadal dysgenesis and an apparently normal female (46,XX) complement was reported in 1960.12 By 1971, a survey of 61 such individuals led to the conclusion that the disorder was inherited in an autosomal recessive fashion.12 A study of all XX gonadal dysgenesis cases in Finland confirmed this conclusion.97
The external genitalia and the streak gonads (see Fig. 90-14) in XX gonadal dysgenesis are indistinguishable from those in gonadal dysgenesis secondary to a sex chromosomal abnormality. Likewise, the endocrine profiles do not differ, but individuals with XX gonadal dysgenesis usually have normal stature (Fig. 90-16). Several pathogenic mechanisms can be postulated, but firm data have not been gathered. Phenocopies for XX gonadal dysgenesis also are well recognized (see Chap. 96).
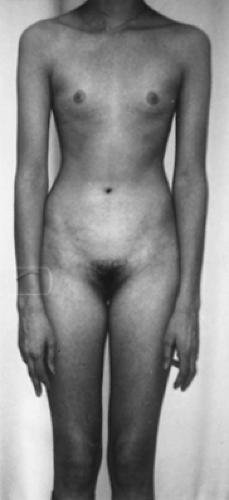 FIGURE 90-16. A case of 46,XX gonadal dysgenesis: a 19-year-old woman with primary amenorrhea. Notice the normal stature and diminished breast development. Circulating gonadotropin levels were markedly elevated. |
Both XX gonadal dysgenesis and neurosensory deafness have occurred in multiple sibs in several families; the occurrence of deaf but fertile male sibs confirms autosomal inheritance.98 The coexistence of gonadal and auditory anomalies probably indicates a syndrome distinct from XX gonadal dys-genesis without deafness (i.e., genetic heterogeneity). Further evidence for genetic heterogeneity can be cited. In several other families, unique patterns of somatic anomalies indicate the existence of mutant genes distinct from those already discussed. These include: XX gonadal dysgenesis and myopathy; XX gonadal dysgenesis and cerebellar ataxia; XX gonadal dysgenesis and metabolic acidosis; XX gonadal dysgenesis, microcephaly, and arachnodactyly.99,100 and 101
Follicle-Stimulating Hormone–Receptor Mutations.
At least one form of XX gonadal dysgenesis is now known to be caused by a mutation of the FSH receptor (FSHR). Affected women present with primary or secondary amenorrhea and elevated serum FSH levels indicative of premature ovarian failure97,102 (see Chap. 96). The mutation originally was identified in a large number of sporadic and familial cases in Finland. Most cases were found in north central Finland, a sparsely populated part of the country. The overall frequency of the disorder in Finland was 1 per 8300 females, a relatively high incidence attributed to a founder effect. The segregation ratio of 0.23 for female sibs was consistent with autosomal recessive inheritance, as was the consanguinity rate of 12%.
Sib-pair analysis using polymorphic DNA markers was used to localize the gene to a specific region of chromosome arm 2p, a region known to contain genes for both FSHR and the LH receptor (LHR). One specific mutation (C566T:alanine to valine) in exon 7 was observed in six multiplex families.102,103 On transvaginal ultrasonography, most of these patients have demonstrable ovarian follicles, raising the possibility of residual receptor activity.103 The C566T mutation was not found in all Finnish XX gonadal dysgenesis patients and is rarely detected in 46,XX women with ovarian failure who reside outside Finland.104,105
Gonadal Dysgenesis in 46,XY Individuals.
Gonadal dysgenesis also can occur in 46,XY individuals. In XY gonadal dys-genesis, affected individuals are phenotypic females who show sexual infantilism and bilateral streak gonads (Fig. 90-17). The gonads may undergo neoplastic transformation (20– 30% prevalence)56 (Fig. 90-18). At least one form of XY gonadal dysgenesis results from an X-linked recessive or male-limited autosomal dominant gene.106,107 Sporadic cases may result from deletion or point mutations within SRY on the Y short arm.14,108,109
Further evidence for genetic heterogeneity lies in the existence of at least three syndromes having XY gonadal dysgenesis as one of their components: XY gonadal dysgenesis and long-limbed camptomelic dwarfism, XY gonadal dysgenesis and ectodermal defects, and the genitopalatocardiac syndrome.36,110,111 and 112
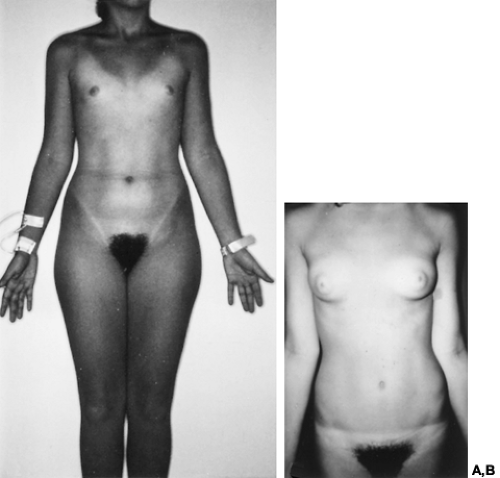 FIGURE 90-17. A and B, Two examples of 46,XY gonadal dysgenesis. Both 16-year-old individuals presented with primary amenorrhea and markedly elevated concentrations of circulating gonadotropins. Both patients had dysgerminoma of an ovary; the patient in B also had a large gonadoblastoma of the contralateral ovary. Breast development as in B is extremely rare and no doubt secondary to hormone production by the patient’s gonadal neoplasm. (B, Photograph reproduced from Villanueva AL, Benirschke K, Campbell J, et al. Complete development of secondary sexual characteristics in a case of 46,XY gonadal dysgenesia. Obstet Gynecol 1984; 64:68S.) |
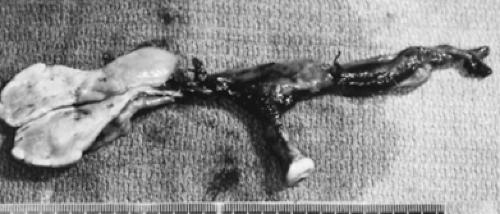 FIGURE 90-18. Gross appearance of the uterus, fallopian tubes, and gonads of 46,XY patient with gonadal dysgenesis. Right gonad (cut) contained dysgerminoma. |
Rudimentary Ovary Syndrome and Unilateral Streak Gonad Syndrome.
The rudimentary ovary syndrome is a poorly defined entity of unknown cause said to be characterized by decreased numbers of follicles. This “syndrome” is heterogeneous, not a single entity. Many cases have been associated with sex chromosomal abnormalities, particularly 45,X/46,XX mosaicism. Similar statements also apply to individuals with the unilateral streak ovary syndrome. For example, a unilateral streak gonad and a contralateral polycystic ovary have been observed in a 46,XX/46,X,i(Xq) individual who became pregnant.113
EVALUATION AND TREATMENT OF GONADAL DYSGENESIS. When Turner stigmata are present, the diagnosis of gonadal dysgenesis usually is made early in childhood. The index of suspicion should be high for any infant with lymphedema of the hands and feet at birth, especially because the somatic anomalies are not very obvious in neonates (see Fig. 90-9). Other children present for evaluation of sexual infantilism, and still others with a male sex-of-rearing may not virilize at the expected age of puberty. Short stature is another common reason that evaluation is sought.
The measurement of circulating gonadotropin concentrations and the determination of the karyotype can establish the
diagnosis. Longitudinal studies have documented elevated gonadotropin levels at all ages in gonadal dysgenesis, indicating absence of appropriate feedback inhibition of the hypothalamic-pituitary unit by the dysgenetic gonads even in childhood.114,115
diagnosis. Longitudinal studies have documented elevated gonadotropin levels at all ages in gonadal dysgenesis, indicating absence of appropriate feedback inhibition of the hypothalamic-pituitary unit by the dysgenetic gonads even in childhood.114,115
Chromosomal studies are indicated to eliminate the possibility of a Y chromosome. The use of the polymerase chain reaction to detect sequences of SRY may well be warranted. If the phenotype and karyotype are compatible, the only tissue needed is blood for lymphocyte culture. If the phenotype and karyotype are incompatible (e.g., tall “45,X” subjects), skin or gonadal fibroblasts also should be cultured to detect any mosaicism. If a Y chromosome is identified, surgical extirpation of the dysgenetic gonads is indicated to prevent neoplasms. Streak gonads usually can be removed by laparoscopy. In appropriate cases in which disseminated malignancies do not involve the gonads, the uterus may be left in situ for donor in vitro fertilization or embryo transfer. The evaluation of other commonly involved organ systems should include a careful physical examination, with special attention to the cardiovascular system, and should include thyroid function tests (including antibodies), fasting blood glucose level, renal function tests, and an intravenous urogram or a renal ultrasonographic scan.
The treatment of individuals with short stature has received significant attention. A multicenter, prospective, randomized trial of administration of GH, alone and in combination with oxandrolone, was initiated in 1983.116,117 Data on 62 girls, who have received 3 to 6 years of treatment, have been published. Given an average height of 143 cm for untreated girls in the United States, the mean height of 151.9 cm in the 30 girls whose therapy was terminated represented a net increase of 8.1 cm (therapy was terminated because the subjects had met study criteria for cessation of treatment, including bone age >14 years and a growth velocity of <2.5 cm over 12 months, or because the subject was satisfied with her current height and elected to discontinue therapy).117
To increase the final height, administration of estrogens in very low doses and anabolic steroids (particularly oxandrolone) has been advocated.117 The success in increasing the final adult height remains arguable, but increased growth in the first 2 or 3 years of therapy without undue acceleration of bone age has been verified.116,118,119 Any increase in the final height appears to be no more than 6 to 8 cm. Although estrogens appear to act by increasing GH secretion, the mode of action of oxandrolone has not been determined. Oxandrolone does not appear to increase the secretion of GH or IGF-I, but it may act directly on growing cartilage. Long-term follow-up suggests that postprandial insulin levels are higher in patients treated with oxandrolone and GH than in patients treated with GH alone.
Treatment strategies that have exogenous GH as their key element are now commonly accepted. An adult height of >150 cm, the widely accepted lower limit of normal height for women in the United States, is now attainable by most girls with this disorder. What the exact dose of GH should be and what additional benefit oxandrolone may contribute are not known. GH administered in doses 25% above those recommended for GH deficiency is proving to be remarkably safe.
To enable the patient to achieve sexual maturation, estrogen therapy should be initiated when the patient is psychologically ready, perhaps at the age of 12 to 13 years, and after GH therapy is completed. Because the aim is to mimic normal pubertal development, therapy is initiated with low-dose estrogen alone (such as oral conjugated estrogens 0.3 mg daily) given continuously for the first several months. A progestogen (5–10 mg of oral medroxyprogesterone acetate or 200 mg of oral micronized progesterone daily for 14 days each month) is then added to prevent the development of endometrial hyperplasia, either when the patient notices vaginal bleeding or after 6 months of unopposed estrogen therapy. Thereafter, the dose of estrogen is increased slowly over 1 to 2 years to daily oral doses of 1.25 to 2.5 mg of conjugated estrogen, 1 to 2 mg of micronized 17β-estradiol, 1.25 to 2.5 mg of piperazine estrone sulfate orally, or 0.05 to 0.1 mg of estradiol transdermally, with the patch changed once or twice weekly depending on the specific preparation. Generally, continued breast tenderness is the first sign that the dose of estrogen is excessive. These patients must be observed especially closely for the development of hypertension with estrogen therapy. The patients and their parents should be informed of the emotional and physical changes that will occur with therapy. Additional principles for estrogen replacement may be found in Chapter 100 (also see ref. 119a).
Individuals with mosaic gonadal dysgenesis and other chromosomal aberrations such as del(X)(p11) or del(X)(q25 to q27) may develop normally at puberty. The decision whether to initiate estrogen therapy can be made on the basis of growth rates, bone age, uterine size determined by ultrasonography, and circulating gonadotropin concentrations. FSH levels in the normal range for age imply functional gonads.
Examination of individuals with sexual infantilism who have been treated with large doses of estrogen (especially conjugated estrogens) from the outset to effect maturation of secondary sexual characteristics often reveals abnormal and inappropriate breast development. The areola often becomes abnormally large, with minimal additional breast tissue. Whether breast contour can be improved in individuals subjected to such inappropriate therapy is not clear.
Pregnancies can now be achieved in these individuals, with success rates of >50%, by using donor oocytes.120 Hormone replacement regimens using transdermal estradiol-17β and intramuscular progesterone have generally been associated with the highest pregnancy rates among those regimens aimed at preparing the endometrium to receive an embryo.
TRIPLE-X SYNDROME (47,XXX). The existence of individuals with a 47,XXX karyotype was first reported in 1959, and an association with premature ovarian failure was noted.121 Since then, study has shown that patients with the triple-X syndrome need not have any impairment in fertility nor any shortening of their reproductive lives.122,123 Premature menopause occurs with increased frequency, however, compared with its incidence in karyotypically normal individuals.124 Moreover, reports of patients with triple-X syndrome associated with immunoglobulin deficiency, together with the finding that the control of T-cell function may be related to the X chromosome, suggest an association between immunologic abnormalities and triple-X syndrome in females with premature ovarian failure.125,126 and 127
Because affected individuals are phenotypically normal, they usually are identified only after presenting with hypergo-nadotropic amenorrhea. Treatment for the premature ovarian failure is the same as it is for menopausal women and is discussed in Chapter 100.
ULLRICH-NOONAN SYNDROME. A syndrome now known as the Ullrich-Noonan or pseudo-Turner syndrome has been described in which phenotypic females present with many of the Turner stigmata and a normal 46,XX chromosomal complement.128,129,131 Moreover, females have functioning ovaries, although the onset of puberty may be delayed. Males also may present with this disorder. The common stigmata include short stature, webbed neck, ptosis, and (unlike in gonadal dysgenesis) right-sided congenital heart disease. Pulmonic stenosis (occurring in perhaps 50% of individuals) and atrial septal defect are the most common cardiac anomalies, although ventricular septal defect, ventricular hypertrophy, patent ductus arteriosus, coarctation of the aorta, and aortic stenosis may be found. Mental retardation, pectus excavatum, cubitus valgus, and lymphedema also may occur (see Chap. 92).
The incidence of this syndrome has been estimated at ˜1 in 8000, with >80% of cases arising from spontaneous mutations.132 Familial clusters consistent with autosomal dominant inheritance have been described.
The existence of this syndrome reinforces the need to perform a karyotype study of individuals presenting with the Turner stigmata. Otherwise, individuals thought to have gonadal dysgenesis may develop normally at puberty and become fertile, much to the surprise of the physician.
SEMINIFEROUS TUBULE DYSGENESIS AND ITS VARIANTS
Klinefelter Syndrome.
Males with at least one Y chromosome and at least two X chromosomes have Klinefelter syndrome.7 Most cases are 47,XXY, but the phenotype may also be associated with 46,XY/47,XXY; 48,XXYY; and 49,XXXXY complements.133,134 and 135 The most characteristic features are seminiferous tubule dysgenesis and androgen deficiency. Somatic anomalies sometimes coexist.133 The presence of a chromosomal abnormality and of elevated gonadotropin levels differentiates Klinefelter syndrome from hypogonadotropic hypogonadism. These conditions are discussed in Chapter 115.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


